

Mott insulator
Mott insulator
Mott insulator

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Material Science<br />
I. d Electron systems<br />
1. Electronic structure of transition-metal ions<br />
(April 9)<br />
2. Crystal structure and band structure (April16)<br />
3. <strong>Mott</strong> <strong>insulator</strong>s (April 23)<br />
4. Metal-<strong>insulator</strong> transition (April 30, May 14)<br />
5. High-temperature superconductivity (May 21)<br />
6. Spin-related phenomena (July 23)<br />
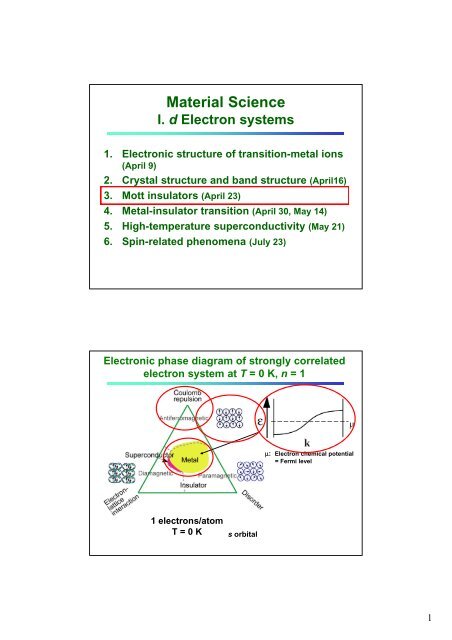
Electronic phase diagram of strongly correlated<br />
electron system at T = 0 K, n = 1<br />
<br />
: Electron chemical potential<br />
= Fermi level<br />
1 electrons/atom<br />
T = 0 K<br />
s orbital<br />
1
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
3.2 Hartree-Fock approximation<br />
3.3 Orbital ordering<br />
3.4 <strong>Mott</strong>-Hubbard vs chargetransfer<br />
type<br />
3.5 Cluster model<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
Hubbard model for single kind of atomic orbitals<br />
(Single-band Hubbard model)<br />
Kinetic energy<br />
Potential energy<br />
= ,<br />
-t<br />
c a + , c a : annihilation, creation operators<br />
t: transfer integral<br />
n a : number operator<br />
U: atomic Coulomb integra<br />
+U<br />
a b a<br />
e.g., d x2-y2 band of CuO 2 plane in high-Tc cuprates<br />
2
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
Super-exchange interaction<br />
-t<br />
X<br />
a b a X<br />
-t<br />
b<br />
Effective spin Hamiltonian = Heisenberg model<br />
Antiferromagnetic<br />
coupling<br />
>0: exchange interaction constant<br />
Various lattices<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
Antiferromagnetic<br />
spin ordering<br />
Frustrated spins<br />
Cu in CuO 2 plane of<br />
high-Tc cuprates<br />
B-site ions of perovskite<br />
lattice<br />
Triangular<br />
lattices<br />
Kagome lattices<br />
B-site ions of spinel<br />
and pyrochlore lattices<br />
3
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
Various lattices<br />
B-site ions of perovskite<br />
lattice<br />
Cu in CuO 2 plane of<br />
high-Tc cuprates<br />
B-site ions of spinel<br />
and pyrochlore lattices<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
U/t 1<br />
(U/2Zt = U/W >> 1)<br />
Metal<br />
<strong>Mott</strong> <strong>insulator</strong><br />
4
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
U/t 1<br />
(U/2Zt = U/W >> 1)<br />
W<br />
<br />
Metal<br />
Density of<br />
states<br />
U<br />
W<br />
<br />
W<br />
Upper<br />
Hubbard<br />
band<br />
E(d 2 )+E(d 0 )-2E(d 1 ) = U<br />
in general E(d <strong>Mott</strong> n+1 <strong>insulator</strong> )+E(d n-1 )-2E(d n ) = U<br />
U<br />
Lower<br />
Hubbard<br />
band<br />
Density of<br />
states<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
3.2 Hartree-Fock approximation<br />
3.3 Orbital ordering<br />
3.4 <strong>Mott</strong>-Hubbard vs chargetransfer<br />
type<br />
3.5 Cluster model<br />
5
2. Crystal structure and band structure<br />
2.5 Magnetically ordered states<br />
Non-magnetic or Pauliparamagnetic<br />
v MF (r)<br />
state<br />
d x2-y2 band on square lattice<br />
Antiferromagnetic state<br />
v MF (r)<br />
= <br />
r<br />
r<br />
2<br />
<br />
= <br />
<br />
Fermi surf<br />
AFM<br />
Brillouin<br />
zone<br />
<br />
k = dx2-y2 - 2t(cos k x a + cos k y a)<br />
2. Crystal structure and band structure<br />
2.5 Magnetically ordered states<br />
Wave function: Bloch orbital<br />
Mean-field Hamiltonian:<br />
(One-electron Hamiltonian)<br />
<br />
Orbital<br />
part<br />
<br />
Nuclei +<br />
core electrons Valence<br />
electrons<br />
<br />
Spin<br />
part<br />
Schroedinger equation<br />
Eigenfunction:<br />
Eigenvalue:<br />
=n: Band index<br />
= ,<br />
- ~ - J H ( - )<br />
6
1. Electronic structure of transition-metal ions<br />
1.3 Coulomb-exchange interaction<br />
Many-electron system<br />
(e.g., 3 electron system)<br />
Expectation<br />
value:U ’ -J ’<br />
<br />
U ”<br />
’<br />
U ’”<br />
”<br />
Coulomb integral<br />
Exchange integral<br />
~ 4-8 eV<br />
Pauli principle ~ 0.5-1 eV<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.2 Hartree-Fock approximation<br />
Wave function: Bloch orbital<br />
Hartree-Fock<br />
Hamiltonian:<br />
(One-electron Hamiltonian)<br />
Hartree-Fock equation<br />
Eigenfunction:<br />
<br />
Potential due to<br />
Orbital<br />
part<br />
Nuclei +<br />
core electrons<br />
Spin<br />
part<br />
Valence<br />
electrons<br />
Exchange<br />
potential<br />
(Non-local)<br />
Eigenvalue:<br />
=n: Band index<br />
= k’’ (U kk’’ - J kk’’ )<br />
=0,1<br />
7
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.2 Hartree-Fock approximation<br />
W<br />
<br />
W<br />
Upper<br />
Hubbard<br />
band<br />
d0 +U<br />
U<br />
<br />
0<br />
Lower d<br />
Hubbard<br />
Density of band<br />
states<br />
<strong>Mott</strong> <strong>insulator</strong><br />
U/t >> 1<br />
Upper<br />
Hubbard<br />
band<br />
W<br />
d0 +U<br />
Density ofU<br />
W<br />
states<br />
<br />
0<br />
Lower d<br />
Hubbard<br />
band<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
3.2 Hartree-Fock approximation<br />
3.3 Orbital ordering<br />
3.4 <strong>Mott</strong>-Hubbard vs chargetransfer<br />
type<br />
3.5 Cluster model<br />
8
Spin, charge, orbital, and lattice degrees of<br />
freedom of strongly correlated electron system<br />
p, d, f orbitals<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
Super-exchange interaction<br />
-t<br />
X<br />
a b a X<br />
-t<br />
b<br />
Effective spin Hamiltonian = Heisenberg model<br />
Antiferromagnetic<br />
coupling<br />
>0: exchange interaction constant<br />
9
orbital1<br />
orbital2<br />
orbital1<br />
orbital2<br />
Super-exchange interaction for degenerate orbitals<br />
-t<br />
X<br />
a<br />
a<br />
-t<br />
-t<br />
-t<br />
b<br />
b<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.3 Orbital ordering<br />
Intermediate-state<br />
energy E =<br />
• 2 nd -order perturbation energy = -2t 2 /E<br />
• U > U’ > U’-J<br />
U<br />
a<br />
U‘ U‘ - J<br />
a<br />
X<br />
-t<br />
b<br />
b<br />
forbidden<br />
Ferro-spin<br />
Antiferro-orbital<br />
1. Electronic structure of transition-metal ions<br />
1.2 Crystal-field splitting<br />
Crystal fields due to anions (e.g., oxygen ions)<br />
Octahedral<br />
coordination<br />
Tetrahedral<br />
coordination<br />
: One-electron energy<br />
( ): Degeneracy (inc. spin)<br />
10Dq ~ 0.5-2 eV<br />
10
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.3 Orbital ordering<br />
W<br />
<br />
W<br />
yz <br />
yz <br />
xy , zx <br />
Ferro-spin, antiferro-orbital ordering in YTiO 3 : Ti 3+ (d 1 )<br />
Upper<br />
Hubbard band<br />
Lower<br />
d0 +U’-J H<br />
U’- J H<br />
d<br />
0<br />
Hubbard band<br />
Density of<br />
states<br />
distorted perovskite structure<br />
Y<br />
Ti<br />
O<br />
W<br />
<br />
W<br />
Upper Hubbard<br />
band<br />
d0 +U’-J H<br />
zx <br />
xy , yz <br />
U’- J H<br />
zx <br />
0<br />
d<br />
Lower Hubbard<br />
band<br />
Density of<br />
states<br />
yz<br />
zx<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.2 Hartree-Fock approximation<br />
Wave function: Bloch orbital<br />
Hartree-Fock<br />
Hamiltonian:<br />
(One-electron Hamiltonian)<br />
Hartree-Fock equation<br />
<br />
Potential due to<br />
Orbital<br />
part<br />
Nuclei +<br />
core electrons<br />
Spin<br />
part<br />
Valence<br />
electrons<br />
Exchange<br />
potential<br />
(Non-local)<br />
Eigenfunction:<br />
Eigenvalue:<br />
Atomic orbitals<br />
=n: Band index<br />
= k’’ (U kk’’ - J kk’’ )<br />
=0,1<br />
11
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
3.2 Hartree-Fock approximation<br />
3.3 Orbital ordering<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer<br />
type<br />
3.5 Cluster model<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
Hubbard model<br />
p-d model<br />
Transition-metal ion (d orbitals)<br />
Non-metal anion (p orbitals)<br />
12
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
Hubbard model<br />
p-d model<br />
=<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
<strong>Mott</strong>-Hubbard-type<br />
<strong>insulator</strong><br />
Charge-transfer-type<br />
<strong>insulator</strong><br />
3d<br />
W<br />
3d<br />
<br />
<br />
3d<br />
W<br />
U <br />
<br />
O 2p<br />
<br />
U<br />
O 2p<br />
Neglected<br />
in Hubbard model<br />
3d<br />
U < <br />
Gap ~ U - W<br />
U > <br />
Gap ~ - W<br />
W: Band width<br />
U : Atomic Coulomb energy (Coulomb integral)<br />
: Charge-transfer enrgy<br />
13
2. Crystal structure and band structure<br />
2.1 What determines crystal structure?<br />
Electronegatigvity ~ [Ionization energy I + Electron affinityA]/2<br />
1.0 1.5<br />
0.8 1.0<br />
Energy<br />
Vacuum level<br />
A<br />
I<br />
1.5 1.6 1.5 1.8<br />
1.2 1.8<br />
2.2<br />
3.5 4.0<br />
2.5<br />
S<br />
2.1<br />
large<br />
small<br />
Number of orthogonal R nl (r)<br />
small<br />
large<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
small<br />
Charge-transfer energy <br />
Valence: 2- 1- …. + 2+ 3+ 4+ 5+<br />
1.0 1.5<br />
large<br />
small<br />
3.5 4.0<br />
large<br />
Transition-metal ions<br />
1.5 1.6 1.5 1.8<br />
2.5<br />
S<br />
0.8 1.0<br />
2.1<br />
1.2<br />
1.8<br />
2.2<br />
large<br />
large<br />
small<br />
Non-TM ions<br />
small<br />
small large<br />
14
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
<strong>Mott</strong>-Hubbard-type<br />
<strong>insulator</strong><br />
Charge-transfer-type<br />
<strong>insulator</strong><br />
3d<br />
W<br />
3d<br />
<br />
<br />
3d<br />
W<br />
U <br />
<br />
O 2p<br />
<br />
U<br />
O 2p<br />
Neglected<br />
in Hubbard model<br />
3d<br />
U < <br />
Gap ~ U - W<br />
U > <br />
Gap ~ - W<br />
W: Band width<br />
U : Atomic Coulomb energy (Coulomb integral)<br />
: Charge-transfer enrgy<br />
2. Crystal structure and band structure<br />
2.1 What determines crystal structure?<br />
Ionic radius<br />
Valence: 2- 1- …. + 2+ 3+ 4+ 5+<br />
large<br />
Units: A<br />
0.74 0.35<br />
1.40 1.33<br />
+ 2+<br />
2- -<br />
1.84 2-<br />
0.67 0.79 0.83 0.69<br />
S<br />
1.38 1.00<br />
+ 2+<br />
3+ 2+ 2+<br />
2+<br />
0.90 0.60 2.24<br />
3+ 6+<br />
2+<br />
2-<br />
0.86<br />
small<br />
small<br />
large<br />
Number of orthogonal R nl (r)<br />
large<br />
small<br />
15
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
large<br />
Atomic Coulomb energy U<br />
Valence: 2- 1- …. + 2+ 3+ 4+ 5+<br />
Units: A<br />
0.74 0.35<br />
1.38 1.00<br />
small large<br />
0.67 0.83 0.69<br />
2+<br />
0.90 0.60<br />
Transition-metal ions<br />
1.40<br />
1.84<br />
2.24<br />
1.33<br />
0.86<br />
small<br />
small<br />
large<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
<strong>Mott</strong>-Hubbard-type<br />
<strong>insulator</strong><br />
Charge-transfer-type<br />
<strong>insulator</strong><br />
3d<br />
W<br />
3d<br />
<br />
<br />
3d<br />
W<br />
U <br />
<br />
O 2p<br />
<br />
U<br />
O 2p<br />
Neglected<br />
in Hubbard model<br />
3d<br />
U < <br />
Gap ~ U - W<br />
U > <br />
Gap ~ - W<br />
W: Band width<br />
U : Atomic Coulomb energy (Coulomb integral)<br />
: Charge-transfer enrgy<br />
16
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
CT<br />
<strong>Mott</strong>-Hubbard vs charge-transfer<br />
Valence: 2- 1- …. + 2+ 3+ 4+ 5+<br />
MH<br />
CT<br />
MH<br />
Transition-metal ions<br />
2+<br />
MH<br />
MT<br />
CT<br />
Non-TM ions<br />
CT<br />
MH<br />
CT<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
Zaanen-Sawatzky-Allen phase diagram<br />
p-band metal<br />
Schematic<br />
Charge-transfer<br />
<strong>insulator</strong><br />
<strong>Mott</strong>-Hubbard<br />
<strong>insulator</strong><br />
d-band metal<br />
Negative<br />
charge-transfer<br />
energy <strong>insulator</strong><br />
p-band metal<br />
Real materials<br />
Charge-transfer<br />
<strong>insulator</strong><br />
<strong>Mott</strong>-Hubbard<br />
<strong>insulator</strong><br />
d-band metal<br />
17
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.1 Hubbard model<br />
3.2 Hartree-Fock approximation<br />
3.3 Orbital ordering<br />
3.4 <strong>Mott</strong>-Hubbard vs chargetransfer<br />
type<br />
3.5 Cluster model<br />
1. Electronic structure of transition-metal ions<br />
1.4 Multiplet splitting<br />
Many-electron system (4 electron system)<br />
3J H -10Dq<br />
3J H<br />
10Dq<br />
n = 4<br />
E: n-electron energy<br />
: one-electon energy<br />
18
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 Cluster model<br />
Spinel-type<br />
structure<br />
<br />
Cluster model<br />
Perovskite-type<br />
structure<br />
Atomic Coulomb energy: U = E(d n+1 )+E(d n-1 )-2E(d n )<br />
Charge-transfer enrgy: = E(d n+1 L)-E(d n )<br />
Transfer integral: T pd = L: Ligand (p) hole<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 Cluster model<br />
Atomic d orbitals<br />
Molecular orbitals consisting of<br />
ligand p orbitals<br />
19
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 Cluster model<br />
Optical absorption spectrum of Ni 2+ (d 8 ) ion in NiO<br />
E – N<br />
Charge-transfer<br />
Optical absorption<br />
d n+1 L<br />
<br />
Ni<br />
O<br />
Optical absorption<br />
Optical<br />
absorption<br />
p-d hybridization Reduction of U, U’, J H<br />
Finite 10Dq<br />
d n<br />
Cluster model CI theory<br />
Ligand-field theory<br />
M. Imada, A. Fujimori and Y. Tokura, Rev. Mod. Phys. 1998<br />
A. Fujimori and F. Minami, PRB 1983<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 Cluster model<br />
CI description of Ni 2+ (d 8 ) ion in NiO<br />
Antibonding states<br />
v<br />
Lu<br />
u<br />
Lv<br />
E – N<br />
6 t pd<br />
d n+1 L<br />
d n <br />
v<br />
u<br />
Hole<br />
p-d hubridization n=8<br />
u = d 3z2-r2 Bonding states Ground state<br />
v = d x2-y2<br />
20
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 <strong>Mott</strong>-Hubbard vs charge-transfer type<br />
<strong>Mott</strong>-Hubbard-type<br />
<strong>insulator</strong><br />
Charge-transfer-type<br />
<strong>insulator</strong><br />
3d<br />
W<br />
3d<br />
<br />
<br />
3d<br />
W<br />
U <br />
<br />
O 2p<br />
<br />
U<br />
O 2p<br />
Neglected<br />
in Hubbard model<br />
3d<br />
U < <br />
Gap ~ U - W<br />
U > <br />
Gap ~ - W<br />
W: Band width<br />
U : Atomic Coulomb energy (Coulomb integral)<br />
: Charge-transfer enrgy<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 Cluster model<br />
A hole doped in the CuO 2 plane: Zhang-Rice singlet<br />
Antibonding states<br />
Hole<br />
v<br />
E – N<br />
6 t pd<br />
d n<br />
U<br />
d n+1 L<br />
v<br />
Lv<br />
S=0<br />
v<br />
Lv<br />
~0.5-1 eV !<br />
p-d hubridization<br />
n=8<br />
Bonding states Ground state<br />
v = d x2-y2<br />
21
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 Cluster model<br />
A hole doped in the CuO 2 plane: Zhang-Rice singlet<br />
O<br />
Hole<br />
Cu<br />
3. <strong>Mott</strong> <strong>insulator</strong>s<br />
3.4 Cluster model<br />
A hole doped in the CuO 2 plane: Zhang-Rice singlet<br />
O<br />
Hole<br />
Cu<br />
22




