Ferroceneâhow it all began
Ferroceneâhow it all began
Ferroceneâhow it all began

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
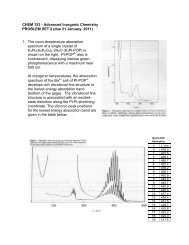
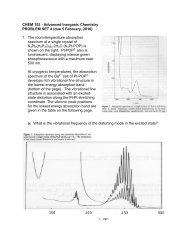
Journal of Organomet<strong>all</strong>ic Chemistry 637–639 (2001) 3–6www.elsevier.com/locate/jorganchemFerrocene—how <strong>it</strong> <strong>all</strong> <strong>began</strong>Peter L. PausonDepartment of Pure and Applied Chemistry, Uniers<strong>it</strong>y of Strathclyde, Thomas Graham Building, Cathedral Street, Glasgow G1 1XL, UKtropolones truly f<strong>it</strong> into this category has remainedcontroversial, they were certainly topical and of muchinterest in this context at the time. My obvious interestsfor my first independent researches were therefore directedtowards further work on e<strong>it</strong>her tropolones orother non-benzenoid systems.(Peter L. Pauson)W<strong>it</strong>h the rest of my family, I had come to the UK in1939 as a refugee from Nazi persecution of the Jews.After completing my secondary education, I enteredGlasgow Univers<strong>it</strong>y in 1942 to study Chemistry. Myinclination towards the organic side was greatly enhancedand my approach to research was moulded bythe inspiring teachings of T.S. Stevens. 1 The offer of aresearch studentship led me to Sheffield Univers<strong>it</strong>y in1946 to do my PhD under the direction of ProfessorR.D. Haworth. 2 My task was to study the chemistry of‘purpurog<strong>all</strong>in’—the brilliant red compound obtainedby oxidation of pyrog<strong>all</strong>ol. We succeeded in establishing<strong>it</strong>s structure as the benzotropolone (1) and showedthat further oxidation followed by decarboxylation ledto -methyltropolone (2), the first synthetic and fullycharacterised simple tropolone.The concept of ‘non-benzenoid aromatics’ was stillrather new and, although the question as to whether1 Discoverer of the Stevens Rearrangement, the McFadyen–Stevens aldehyde synthesis, and the Bamford–Stevens reaction.2Best remembered for the Haworth phenanthrene synthesis.On completion of my degree, I had moved toDuquesne Univers<strong>it</strong>y 3 in 1949. There, I read w<strong>it</strong>h interest,but also scepticism, R.D. Brown’s paper (Nature,1950), which suggested that fulvalene (3) might showaromatic properties. The ch<strong>all</strong>enge to e<strong>it</strong>her prove or3 This Catholic univers<strong>it</strong>y in P<strong>it</strong>tsburgh was better known for <strong>it</strong>sbasketb<strong>all</strong> team than for <strong>it</strong>s academic achievements. Its chemistrystudents were mostly pre-meds, but <strong>it</strong> offered BS and MS degrees inchemistry although not yet PhD. I had applied in response to anadvertisement in Chemical and Engineering News and, after their firstselected candidate w<strong>it</strong>hdrew at short notice, I was offered the post ofAssistant Professor w<strong>it</strong>hout interview.0022-328X/01/$ - see front matter © 2001 Published by Elsevier Science B.V.PII: S0022-328X(01)01126-3
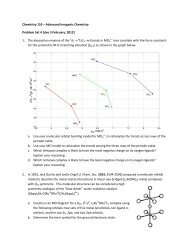
4P.L. Pauson / Journal of Organomet<strong>all</strong>ic Chemistry 637–639 (2001) 3–6disprove his suggestion was heightened by my belief thatthe compound might be accessible in just two steps: thecoupling of two molecules of cyclopentadienylmagnesiumbromide to give the dihydro compound 4, followedby dehydrogenation. This, therefore, was the project Ioffered to Tom Kealy when he expressed an interest, in1951, in working w<strong>it</strong>h me for his MS degree.The reductive coupling of Grignard reagents RMgXby trans<strong>it</strong>ion metal halides to give hydrocarbons RR isa general reaction that is known to be particularly easywhen R is an <strong>all</strong>ylic group. Although other halides, e.g.CoCl 2 , were regarded as better, we chose FeCl 3 for tworeasons: (i) It is a common and ether-soluble reagent in<strong>it</strong>s anhydrous form, whereas <strong>all</strong> the other candidateswould have had to be made from hydrates; and (ii) iffulvalene were truly aromatic, the oxidising power ofFe(III) might suffice to effect the dehydrogenation stepand thus give us fulvalene in a one-pot process.(T.J. Kealy)Tom and I did the crucial experiment together one dayin July 1951: making EtMgBr in ether, at the same timecracking the dimer of and redistilling cyclopentadiene,which was then added to the Grignard solution todisplace ethane and generate cyclopentadienylmagnesiumbromide, fin<strong>all</strong>y adding the FeCl 3 . Standardworkup of this mixture by pouring onto ice and ammoniumchloride produced a yellow ether layer—not theyellow aqueous layer expected if the iron(III) had remainedunreduced. Moreover, a few drops of the yellowsolution that had escaped onto the outside of theseparating funnel evaporated, leaving yellow crystals.Could we have made a yellow hydrocarbon, perhapseven the desired fulvalene? It seemed too good to be true.We soon had a batch of purified crystals and, notwishing to imply that the results should add up to 100%,these were sent for C,H microanalysis as containing C,H, and O. When the results came back a few days later,the large defic<strong>it</strong> from 100% clearly required an elementheavier than oxygen and <strong>it</strong> needed l<strong>it</strong>tle ar<strong>it</strong>hmetic tofind that these results accurately f<strong>it</strong>ted C 10 H 10 Fe. Wenow had to analyse for iron, both qual<strong>it</strong>atively andquant<strong>it</strong>atively. The compound dissolved in strong sulphuricacid but was largely recovered unchanged ondilution. We had to boil w<strong>it</strong>h strong n<strong>it</strong>ric acid before wecould get the standard qual<strong>it</strong>ative tests for iron to work,and even this was insufficient to give accurate quant<strong>it</strong>ativeresults. These were only obtained after fuming todryness w<strong>it</strong>h HClO 4 .What was this remarkable substance we had made?We were well aware of the common belief that bondsbetween trans<strong>it</strong>ion metals and hydrocarbon groupswould always be unstable—based on the failure of <strong>all</strong>recorded attempts to form such links. Yet, here we hada compound, formed from two C 5 H 5 un<strong>it</strong>s and an ironatom, which was not only isolable but also stable to hightemperatures and unaffected by water and by bothstrong acids and alkalis. True, we knew of the exceptionsto the <strong>all</strong>eged non-existence of organo-trans<strong>it</strong>ion metalcompounds: Zeise (1827) had made K[PtCl 3 (C 2 H 4 )] andsome methylplatinum compounds had been described—but platinum was seen as ‘atypical’; Hein (1919) haddescribed ‘polyphenylchromium salts’ and Reihlen(1930) had reacted iron pentacarbonyl w<strong>it</strong>h butadiene togive C 4 H 6 Fe(CO) 3 . These substances tended to be ignoredsimply because their structures were unknown.Whatever our product might be, <strong>it</strong> was clearly somethingvery novel and unexpected, and we thereforereported our findings in a note to Nature—despatchedw<strong>it</strong>hin a week of getting that C,H analysis. We wrote thestructure as 5.I attended the large IUPAC Congress in New York acouple of months later and there handed a sample of ourcrystals to J.M. Robertson, the distinguished X-raycryst<strong>all</strong>ographer who was Professor of Physical Chemistryat Glasgow Univers<strong>it</strong>y.Soon several people independently guessed the correctstructure. The first of these was W. von E. Doering,whom I vis<strong>it</strong>ed at Columbia Univers<strong>it</strong>y immediatelyafter the IUPAC Congress and told about our finding.Unfortunately, I failed to understand his suggestion,which included the possibil<strong>it</strong>y that magnetic susceptibil<strong>it</strong>ymight provide evidence; I was too reluctant to exposemy ignorance and my lack of understanding and wascontent to awa<strong>it</strong> the defin<strong>it</strong>ive answer expected fromX-ray work. In the meantime, through circumstancesbeyond his control, 4 no results were forthcoming fromRobertson before several others undertook X-ray structuralwork after our note appeared in Nature. Ironic<strong>all</strong>y,one of these was Jack Dun<strong>it</strong>z, next to whom I was s<strong>it</strong>tingat the IUPAC meeting while listening to Robertson’stalk and wa<strong>it</strong>ing to give him the sample.4 Robertson was on his way to Cornell Univers<strong>it</strong>y as that year’s‘Baker Lecturer’ and preoccupied w<strong>it</strong>h wr<strong>it</strong>ing the book which wasexpected of holders of this vis<strong>it</strong>ing appointment.
P.L. Pauson / Journal of Organomet<strong>all</strong>ic Chemistry 637–639 (2001) 3–6 5In those days, the Chemical Society published lists ofpapers accepted for publication in <strong>it</strong>s journal. About amonth before our Nature note was due, I read of apaper on ‘Dicyclopentadienyliron’ by Miller, Tebbothand Tremaine of the Br<strong>it</strong>ish Oxygen Co. I wrote to Dr.Miller and told him that we appeared to have the samecompound, and so <strong>it</strong> proved when he sent me a proofcopy of the J. Chem. Soc. paper. It also showed thatthey had used a radic<strong>all</strong>y different route and that theirpaper had been subm<strong>it</strong>ted before we ever tried thereaction, but <strong>it</strong> was to appear in the February 1952issue of J. Chem. Soc., whereas Nature’s faster methodof publication gave us the apparent precedence w<strong>it</strong>h a1951 publication date. In an accompanying note Millermentioned that they had had the compound for about3 years. Even this, however, was qu<strong>it</strong>e possibly not thefirst preparation. The following tale was told about ayear later by E.O. Brimm of Linde Air Products.Brimm, who had done some work on metal carbonylsand other organomet<strong>all</strong>ics, was one of several industrialchemists who, on reading our report, wanted to makesome of the compound. Therefore he enquired of acolleague at Union Carbide (Linde’s parent company)whether they had any cyclopentadiene. The reply thatthey no longer did so was accompanied by the statementthat, some years previously, they had terminatedwork on the cracking of cyclopentadiene because ayellow sludge clogged the iron pipes they used. Theyhad not attempted to isolate or analyse this but hadkept a bottle of <strong>it</strong>; <strong>it</strong> was ferrocene. The Br<strong>it</strong>ish Oxygengroup’s synthesis was, in fact, not dissimilar as <strong>it</strong> involvedpassing cyclopentadiene over a heated, iron-containingammonia synthesis catalyst.As is well-known, the correct structure of dicyclopentadienylironwas put forward in two independent publications:one by E.O. Fischer and W. Pfab, the other byG. Wilkinson, M. Rosenblum, M.C. Wh<strong>it</strong>ing, and R.B.Woodward. Both groups had repeated our preparationand made physical measurements that strongly supportedthe ‘Doppelkegel’ or ‘sandwich’ structure butfell short of full proof. Fischer relied chiefly on preliminaryX-ray data indicating that the molecules arecentrosymmetric, while the Harvard group c<strong>it</strong>ed thesingle C–H frequency in the IR and the diamagnetism.Only weeks went by before two independent completeX-ray structure determinations were published. 5At Woodward’s suggestion that the compound mightbe aromatic, Wh<strong>it</strong>ing and Rosenblum went on to showthat <strong>it</strong> readily undergoes Friedel–Crafts acylation and,on this basis, suggested the name ‘ferrocene’—w<strong>it</strong>h the‘ene’ ending implying aromatic<strong>it</strong>y. We had indeed prepareda novel, non-benzenoid aromatic, but a veryunexpected add<strong>it</strong>ion to that class!5By Dun<strong>it</strong>z and Orgel and by Eiland and Pepinsky.Both Fischer and Wilkinson very soon started addingbis-cyclopentadienyls of other trans<strong>it</strong>ion metals; TomKealy had tried nickel and cobalt but was thwarted bythe insolubil<strong>it</strong>y of their anhydrous halides, which preventedtheir reaction w<strong>it</strong>h the ethereal solution of cyclopentadienylmagnesiumbromide. Fischer overcamethis problem by turning to the hexammine complexes ofthese metals and Wilkinson by using the acetylacetonates.In the meantime I was spending the academic year1951–1952 at the Univers<strong>it</strong>y of Chicago as a postdoctoralfellow working on peroxide chemistry w<strong>it</strong>h M.S.Kharasch. My next move was to Harvard where I wasappointed to a ‘Du Pont Fellowship’ to work independently.Although my application for this had beenbased on a scheme to synthesise the alkaloid colchicine(a tropolone), Professor Bartlett, as Chairman of thedepartment, made clear that I was free to do whateverI liked. It cannot have been unexpected that by then Iwas anxious to play a part in the work on met<strong>all</strong>oceneswhich was in full swing in both Woodward’s andWilkinson’s laboratories.En route to Harvard, I vis<strong>it</strong>ed the Du Pont laboratoriesat Wilmington. There, Dr Victor Weinmayr wasdoing work on the organic chemistry of ferrocene. Ontalking about other metals, he told me that nickelworked and that I should feel free to use this informationprovided that I did not disclose <strong>it</strong>s source.Combining this hint w<strong>it</strong>h what I knew already, I thereforeemployed nickel acetyl acetonate in my first experimentsat Harvard and, by the time Wilkinson andCotton both returned from their summer vacations, Iwas able to show them a sample of the beautifulemerald-green crystals of nickelocene. They showedthat <strong>it</strong> is paramagnetic and we extended our jointstudies to the benzo derivatives of ferrocene and of thecobaltocenium cation, which I prepared from indene bysimilar techniques, leaving them to do <strong>all</strong> the physicalmeasurements.During this and the following 2 years, Fischer andWilkinson made bis-cyclopentadienyl–metal and cyclopentadienylmetalcarbonylcompounds of most of thetrans<strong>it</strong>ion metals. It seemed almost inev<strong>it</strong>able that theywere in constant compet<strong>it</strong>ion as to who would be firstw<strong>it</strong>h the next, fairly obvious target. 6At the end of my year at Harvard, I returned to theUK to take up a lectureship in organic chemistry atSheffield Univers<strong>it</strong>y. There, I soon had the benef<strong>it</strong> ofPhD students to do <strong>all</strong> the hard work for me. Weconcentrated on the organic aspects. The first student(G.D. Broadhead), after showing that ferrocene undergoesacetylation very much faster than anisole, foundthat <strong>it</strong> would undergo formylation under Vilsmeyer6 This compet<strong>it</strong>ion effectively ceased in 1955, when Fischer’s success(w<strong>it</strong>h W. Hafner) in making bis-benzenechromium led him toconcentrate heavily on arene complexes.
6P.L. Pauson / Journal of Organomet<strong>all</strong>ic Chemistry 637–639 (2001) 3–6cond<strong>it</strong>ions (as also shown independently by Rosenblum)and then tried azo-coupling, thereby finding theunexpected arylation. The second student (B.F. H<strong>all</strong>am),after some work on cyclopentadiene–iron carbonylcomplexes, took on the task of verifying mybelief that Reihlen’s above-mentioned butadienetricarbonylironis also a -complex. By repeating <strong>it</strong>s preparationand also making the cyclohexadiene complex, heopened up the whole field of olefin, diene, and trienecomplexes—but that is the beginning of a larger storybeyond the scope of the present topic.It was undoubtedly the availabil<strong>it</strong>y of good methodsof structure determination coupled w<strong>it</strong>h the greatertheoretical understanding which made possible the explosivegrowth of interest in organo-trans<strong>it</strong>ion metalchemistry following directly from the discovery of ferrocene,whereas the earlier findings had led nowhere.Person<strong>all</strong>y, I feel extremely lucky to have been involvedfrom the start and I owe a huge debt of grat<strong>it</strong>ude to asuccession of co-workers who made <strong>it</strong> possible for meto participate w<strong>it</strong>h great enjoyment in these developmentsthroughout these 50 years.