Caractéristiques spectrales des composés aromatiques
Caractéristiques spectrales des composés aromatiques
Caractéristiques spectrales des composés aromatiques

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
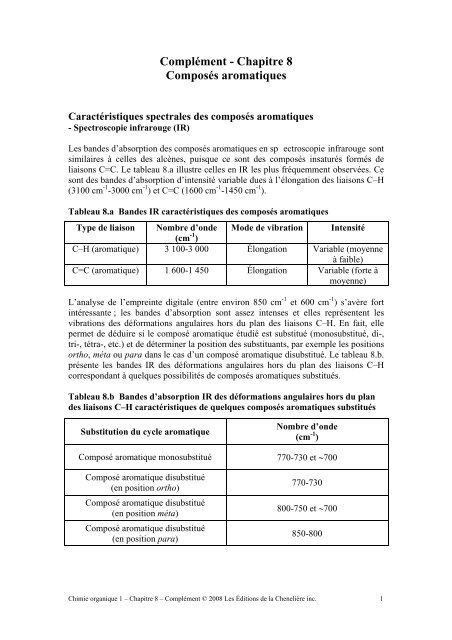
Complément - Chapitre 8Composés <strong>aromatiques</strong>Caractéristiques <strong>spectrales</strong> <strong>des</strong> composés <strong>aromatiques</strong>- Spectroscopie infrarouge (IR)Les ban<strong>des</strong> d’absorption <strong>des</strong> composés <strong>aromatiques</strong> en sp ectroscopie infrarouge sontsimilaires à celles <strong>des</strong> alcènes, puisque ce sont <strong>des</strong> composés insaturés formés deliaisons C=C. Le tableau 8.a illustre celles en IR les plus fréquemment observées. Cesont <strong>des</strong> ban<strong>des</strong> d’absorption d’intensité variable dues à l’élongation <strong>des</strong> liaisons C–H(3100 cm -1 -3000 cm -1 ) et C=C (1600 cm -1 -1450 cm -1 ).Tableau 8.a Ban<strong>des</strong> IR caractéristiques <strong>des</strong> composés <strong>aromatiques</strong>Type de liaison Nombre d’onde Mode de vibration Intensité(cm -1 )C–H (aromatique) 3 100-3 000 Élongation Variable (moyenneà faible)C=C (aromatique) 1 600-1 450 Élongation Variable (forte àmoyenne)L’analyse de l’empreinte digitale (entre environ 850 cm -1 et 600 cm -1 ) s’avère fortintéressante ; les ban<strong>des</strong> d’absorption sont assez intenses et elles représentent lesvibrations <strong>des</strong> déformations angulaires hors du plan <strong>des</strong> liaisons C–H. En fait, ellepermet de déduire si le composé aromatique étudié est substitué (monosubstitué, di-,tri-, tétra-, etc.) et de déterminer la position <strong>des</strong> substituants, par exemple les positionsortho, méta ou para dans le cas d’un composé aromatique disubstitué. Le tableau 8.b.présente les ban<strong>des</strong> IR <strong>des</strong> déformations angulaires hors du plan <strong>des</strong> liaisons C–Hcorrespondant à quelques possibilités de composés <strong>aromatiques</strong> substitués.Tableau 8.b Ban<strong>des</strong> d’absorption IR <strong>des</strong> déformations angulaires hors du plan<strong>des</strong> liaisons C–H caractéristiques de quelques composés <strong>aromatiques</strong> substituésSubstitution du cycle aromatiqueComposé aromatique monosubstituéComposé aromatique disubstitué(en position ortho)Composé aromatique disubstitué(en position méta)Composé aromatique disubstitué(en position para)Nombre d’onde(cm -1 )770-730 et ∼700770-730800-750 et ∼700850-800Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 1
On constate au tableau 8.b que les ban<strong>des</strong> IR <strong>des</strong> liaisons C–H au niveau del’empreinte digitale sont relativement près les unes <strong>des</strong> autres. Les spectresinfrarouges sont souvent difficiles à interpréter. De ce fait, il faut effectuer plusieurscaractérisations (RMN 1 H, spectre de masse…) d’un composé étudié, en plus duspectre infrarouge, pour pouvoir apporter <strong>des</strong> conclusions avec certitude quant à sanature.Dans les spectres IR du benzène et du o-xylène illustrés à la figure 8.a, les ban<strong>des</strong> IR<strong>des</strong> composés <strong>aromatiques</strong> caractéristiques sont observées.Figure 8.a Spectres IRa) du benzèneSpectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 2
) du o-xylèneSpectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.c) du m-xylèneSpectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 3
d) et du p-xylèneSpectre réalisé sur Thermo Electronic Corporation, Nicolet IR 100par Danielle Lapierre et Claude Gaudreault du Collège Jean-de-Brébeuf.- Résonance magnétique nucléaire (spectre RMN)Le cône d’anisotropie crée, dans le cas du cycle benzénique, un très grand déblindageau niveau <strong>des</strong> hydrogènes directement liés au cycle. Ceux-ci résonnent alors à unchamp très faible et ont donc <strong>des</strong> déplacements chimiques élevés, soit entre 6,6 ppmet 8,0 ppm. Par exemple, les hydrogènes du benzène, sans substituant, possèdent undéplacement chimique de 7,27 ppm. Leur signal est un singulet, les six hydrogènesétant équivalents (voir la figure 8.b).Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 4
Figure 8.b Spectre RMN 1 H du benzène (solvant : CDCl 3 )Spectre réalisé sur Bruker Avance 400 PJAB par François Raymond, étudiantau doctorat au laboratoire du professeur James D. Wuest à l'Université de Montréal.Les spectres RMN 1 H <strong>des</strong> composés <strong>aromatiques</strong> sont souvent assez complexes àanalyser. Une de leur particularité est qu’ils révèlent <strong>des</strong> couplages entre <strong>des</strong>hydrogènes plutôt éloignés. On les distingue les uns <strong>des</strong> autres par leurs constantes decouplage (J) d’intensité décroissante. Ainsi, les hydrogènes en position ortho ont <strong>des</strong>constantes de couplage variant entre 6 Hz et 8 Hz et ceux en position méta en ontd’environ 1 Hz à 3Hz. Les constantes de couplage <strong>des</strong> hydrogènes en position parasont très faibles, voire inexistantes, allant de 0 Hz à 1 Hz.De plus, les substituants activants (qui enrichissent le cycle aromatique en électrons)ont un effet blindant tandis que les substituants désactivants (qui appauvrissent lecycle aromatique en électrons) ont un effet déblindant sur les déplacements chimiques<strong>des</strong> hydrogènes benzéniques. Les hydrogènes situés en position ortho du substituantsubissent davantage l’effet de ce dernier. Le tableau 8.c présente une approximationde l’effet <strong>des</strong> différents substituants sur les hydrogènes benzéniques en fonction deleur position. Les hydrogènes d’un cycle benzénique ne seront donc pas touséquivalents selon la nature et le nombre de substituants sur le cycle, ce qui nécessitealors une analyse plus approfondie, les spectres étant plus compliqués.Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 5
Tableau 8.c* Influence de la nature de divers substituants sur les hydrogènesbenzéniques d’un composé aromatique monosubstitutéSubstituant Position ortho Position méta Position para-CH 3 , -R -0,15 -0,1 -0,1-CH=CH 2 +0,2 +0,2 +0,2-COOH, -COOR +0,8 +0,15 +0,2-CN +0,3 +0,3 +0,3-CONH 2 +0,5 +0,2 +0,2-COR +0,6 +0,3 +0,3-SR +0,1 -0,1 -0,2-NH 2 , -NHR -0,8 -0,15 -0,4-N(CH 3 ) 2 -0,5 -0,2 -0,5-I +0,3 -0,2 -0,1-CHO +0,7 +0,2 +0,4-Br 0 0 0-NHCOR +0,4 -0,2 -0,3-Cl 0 0 0+-NH 3 +0,4 +0,2 +0,2-OR -0,2 -0,2 -0,2-OH -0,4 -0,4 -0,4-OCOR +0,2 -0,1 -0,2-NO 2 +1,0 +0,3 +0,4-SO 3 H, -SO 2 Cl,-SO 2 NH 2 , etc.+0,4 +0,1 +0,1*Données tirées du manuel de W. Kemp, Organic Spectroscopy, Third Edition, W.H. Freeman andCompany, New York, 1995.Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 6
Ainsi, les déplacements chimiques <strong>des</strong> hydrogènes du 4-bromo-2-méthylanilineNH 2H 1NH 2 en position ortho(fortement blindant)H 1 : 7,27 + (-0,1) + (-0,8) = 6,37 ppmH 3BrH 2Méthyle en position méta(faiblement blindant)NH 2 en position métaNH 2 en position métaH 2 : 7,27 + (-0,1) + (-0,15) = 7,02 ppmMéthyle en position paraH 3 : 7,27 + (-0,15) + (-0,15) = 6,97 ppmMéthyle en position orthoN.B. Le Br n'a pas d'influencesur les déplacements chimiques<strong>des</strong> hydrogènes peut importe saposition sur le cycle.Remarque : Les valeurs fournies au tableau 8.c sont fiables, mais demeurent uneapproximation. Les déplacements chimiques calculés ne sont donc pas les valeursexactes obtenues dans un spectre RMN. À titre d’exemple, les calculs théoriquesprésentés ci-haut permettent de prévoir grossièrement les déplacements chimiques <strong>des</strong>différents hydrogènes du 4-bromo-2-méthylaniline, mais ce ne sont pas exactementles valeurs analysées sur le spectre RMN de ce composé illustré à la figure 8.c. Onobserve en effet une inversion <strong>des</strong> déplacements chimiques <strong>des</strong> hydrogènes H 2 et H 3 .Par conséquent, pour réaliser une bonne analyse, il faut non seulement se fier auxdéplacements chimiques calculés, mais il est important de toujours considérer lesconstantes de couplage.Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 7
Figure 8.c Spectre RMN 1 H du 4-bromo-2-méthylaniline (solvant : CDCl 3 )Spectre réalisé sur Bruker Avance 400 PJAB par François Raymond, étudiantau doctorat au laboratoire du professeur James D. Wuest à l'Université de Montréal.Pour ce qui est <strong>des</strong> spectres RMN 13 C, les carbones du cycle benzénique possèdent<strong>des</strong> déplacements chimiques entre 120 ppm et 135 ppm, lorsqu’ils n’ont pas <strong>des</strong>ubstituants (voir la figure 8.d). En effet, ces derniers, selon leur nature, affectent lesdéplacements chimiques.Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 8
Figure 8.d Spectre RMN 13 C du benzène (solvant : CDCl 3 )Spectre réalisé sur Bruker Avance 400 PJAB par François Raymond, étudiantau doctorat au laboratoire du professeur James D. Wuest à l'Université de Montréal.Chimie organique 1 – Chapitre 8 – Complément © 2008 Les Éditions de la Chenelière inc. 9