cej zmiany stężenia detektora lub produktu jego reakcji w czasie (rys. 4). Toprzesunięcie spowodowane jest tym, że antyoksydanty zawarte w próbcenie dopuszczają do reakcji rodnika z detektorem. Stąd podstawowym wymogiemtej metody jest, aby reakcja próbki z rodnikiem była znacznie szybszaod analogicznej reakcji detektora.Duża ilość metod pomiaru CPA powoduje, że ten sam materiał mierzonyza pomocą różnych metod może dawać odmienne wyniki. W celuujednolicenia wyników w większości przypadków stosuje się przeliczanie ichna jeden ze standardów, przeważnie stężenie Troloksu, syntetycznego, rozpuszczalnegow wodzie analogu witaminy E.CHROMATOGRAFICZNY POMIAR STRESU OKSYDACYJNEGOCPA jest odwrotnie skorelowany z inną sumaryczną wielkością, mianowicieze stresem oksydacyjnym. Pośrednią grupę technik oznaczaniastresu oksydacyjnego jest analiza niektórych związków naturalnie występującychw układach biologicznych. Zaliczyć do tego można oznaczanie sumarycznegostężenia tioli, stosunku stężeń glutationu do jego utlenionej postaciczy kwasu askorbinowego do dehydroaskorbinowego. Wolne rodniki reagująz praktycznie wszystkimi związkami biologicznie aktywnymi (najbardziejistotne są ich oddziaływania z DNA, białkami i lipidami). Produkty tych reakcjisłużą do wyznaczenia stopnia uszkodzenia tych związków. Ponieważ sąone nietrwałe ich bezpośrednie oznaczanie w próbkach materiału biologicznego(np. w osoczu, czy w homogenatach tkankowych) nie jest zazwyczajmożliwe, dlatego też pośrednią metodą oznaczania stresu oksydacyjnegojest analiza produktów tych reakcji. Uszkodzenie wolnorodnikowe białekmierzone jest stopniem przereagowania tyrozyny w bityrozynę lub stężeniemgrup karbonylowych. Znanych jest ponad 20 wolnorodnikowych uszkodzeńzasad purynowych i pirymidynowych. Wśród istotnych wskaźników uszkodzeniakwasów nukleinowych wyróżnić należy 8-okyguaninę i 8-oksyadeninęi 8-hydroksy-2-deoksyguanozynę. Inne podejście polega na określeniu aktywnościenzymu reperującego nici DNA, poli-ADP-rybozy. Wydaje się, żew chwili obecnej największe zainteresowanie wzbudza określenie stopniaperoksydacji lipidów. Oznaczanymi produktami ich peroksydacji jest dialdehydmalonowy (MDA) [7], 4-hydroksy-2-nonenal (HNO), oksysterole (7α-,7β-, 24, 25- i 27-hydroksycholesterol oraz epoksycholesterol [8a]), hydroksynadtlenki,sprzężone dieny czy też ostatnio intensywnie badane izomeryprostaglandyny F 2 . F 2 -izoprostany (w szczególności 8-epiPGF 2α ) powstająprzede wszystkim w procesie peroksydacji kwasu arachidonowego i eikozanoidów(prostoglandyny, leukotrieny i tromboksany).Produktami peroksydacji, głównie wielonienasyconych kwasów tłuszczowych(PUFA), są aldehydy, hydroksynadtlenki i ketony, a ich produktemkońcowym jest dialdehyd malonowy (MDA). Pomiar jego stężenia jest powszechnieuznawany za miarę wolnorodnikowego uszkodzenia lipidów [7].Najczęściej stosuje się do tego, ze względu na prostotę obsługi i wysoką54
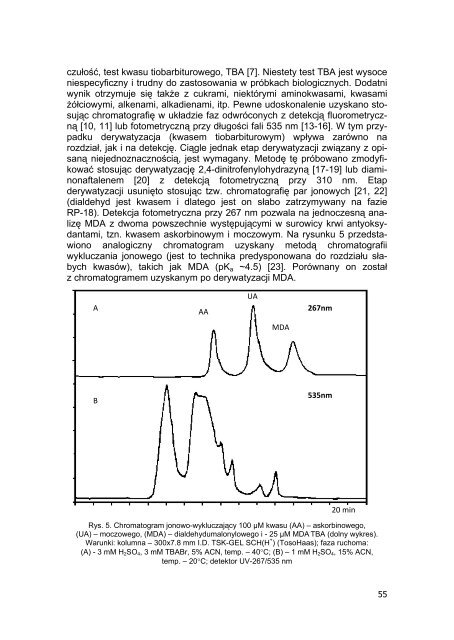
czułość, test kwasu tiobarbiturowego, TBA [7]. Niestety test TBA jest wysoceniespecyficzny i trudny do zastosowania w próbkach biologicznych. Dodatniwynik otrzymuje się także z cukrami, niektórymi aminokwasami, kwasamiżółciowymi, alkenami, alkadienami, itp. Pewne udoskonalenie uzyskano stosującchromatografię w układzie faz odwróconych z detekcją fluorometryczną[10, 11] lub fotometryczną przy długości fali 535 nm [13-16]. W tym przypadkuderywatyzacja (kwasem tiobarbiturowym) wpływa zarówno narozdział, jak i na detekcję. Ciągle jednak etap derywatyzacji związany z opisanąniejednoznacznością, jest wymagany. Metodę tę próbowano zmodyfikowaćstosując derywatyzację 2,4-dinitrofenylohydrazyną [17-19] lub diaminonaftalenem[20] z detekcją fotometryczną przy 310 nm. Etapderywatyzacji usunięto stosując tzw. chromatografię par jonowych [21, 22](dialdehyd jest kwasem i dlatego jest on słabo zatrzymywany na fazieRP-18). Detekcja fotometryczna przy 267 nm pozwala na jednoczesną analizęMDA z dwoma powszechnie występującymi w surowicy krwi antyoksydantami,tzn. kwasem askorbinowym i moczowym. Na rysunku 5 przedstawionoanalogiczny chromatogram uzyskany metodą chromatografiiwykluczania jonowego (jest to technika predysponowana do rozdziału słabychkwasów), takich jak MDA (pK a ~4.5) [23]. Porównany on zostałz chromatogramem uzyskanym po derywatyzacji MDA.AAAUA267nmMDAB535nm20 minRys. 5. Chromatogram jonowo-wykluczający 100 μM kwasu (AA) – askorbinowego,(UA) – moczowego, (MDA) – dialdehydumalonylowego i - 25 μM MDA . TBA (dolny wykres).Warunki: kolumna – 300x7.8 mm I.D. TSK-GEL SCH(H + ) (TosoHaas); faza ruchoma:(A) - 3 mM H 2 SO 4 , 3 mM TBABr, 5% ACN, temp. – 40°C; (B) – 1 mM H 2 SO 4 , 15% ACN,temp. – 20°C; detektor UV-267/535 nm55
- Page 1 and 2:
POSTĘPY CHROMATOGRAFIIPraca zbioro
- Page 3: SPIS TREŚCIPrzedmowa..............
- Page 7 and 8: Piotr M. SŁOMKIEWICZ 1 , Zygfryd W
- Page 9 and 10: W komorze 2 znajduje się pojemnik
- Page 11 and 12: czyna przypływać przez komorę 2
- Page 13 and 14: 4Cnapięcie [mV]3Rys. 6. Chromatogr
- Page 15 and 16: Bronisław K. GŁÓD 1 , Paweł PIS
- Page 17 and 18: Rys. 1. Metabolizm dopaminyL-dopa p
- Page 19 and 20: Dopamina, której niedobór w czę
- Page 21 and 22: katecholowej oraz kompleksowaniu ż
- Page 23 and 24: typu skonstruowano w Instytucie Ele
- Page 25 and 26: takich jak zmęczenie czy stres. Dr
- Page 27 and 28: CPA SUROWICY KRWI PACJENTÓW Z CHOR
- Page 29 and 30: mo wyższych stężeń w surowicy e
- Page 31: LITERATURA1. C. Courderot-masuyer,
- Page 34 and 35: dopuszczające leki jak np. FDA nie
- Page 36 and 37: W przypadku przeprowadzania analizy
- Page 38 and 39: pompaHPLCkolumna chiralnakolumnarea
- Page 40 and 41: 25. K. Lorenz, E. Yashima, Y. Okamo
- Page 42 and 43: Tlen, chociaż jest konieczny do ż
- Page 44 and 45: σ2∗pπ * 2pπ 2pσ 2psingletowy
- Page 46 and 47: Rozpuszczalność tlenku azotu w wo
- Page 48 and 49: nia wolnych rodników. Dlatego nie
- Page 50 and 51: na do nich również poliaminy biog
- Page 52 and 53: trwały rodnik, możliwy do oznacze
- Page 56 and 57: Rodniki hydroksylowe są wyjątkowo
- Page 58 and 59: COOHOCOCH3aspirynahydrolizaCOOHCOC
- Page 60 and 61: W statycznej, chromatograficznej me
- Page 62 and 63: sowej. Warto zwrócić w tym przypa
- Page 64 and 65: 35. W. Pakszys, B.K. Głód, P.P. L
- Page 66 and 67: 95. A. Ghiselli, M. Serafini, F. Na
- Page 68 and 69: nych umożliwiających oznaczanie f
- Page 70 and 71: W monitoringu wód powierzchniowych
- Page 72: RT: 4.01 - 19.97IntensityIntensity1
- Page 75 and 76: Ciekawą rolę pełnią zbiorniki w
- Page 77: Stosowana w badaniach procedura ana
- Page 80 and 81: wania substancji, zwłaszcza, w ska
- Page 82 and 83: - Kolumnowa, elucyjna chromatografi
- Page 84 and 85: Przykłady a, a’, b, b’, c, c
- Page 86 and 87: ZJAWISKA PRZEŁADOWANIA KOLUMNY (PO
- Page 88 and 89: ZASADY OPTYMALNEGO STOSOWANIA PREPA
- Page 90 and 91: - przeładowanie objętościoweW ch
- Page 92 and 93: Średnica kolumnyNajbardziej celowe
- Page 94 and 95: Średnica ziaren wypełnienia kolum
- Page 96 and 97: RetencjaSilna retencja składników
- Page 98 and 99: OPERACJE JEDNOSTKOWE I TECHNOLOGIA
- Page 100 and 101: Rys. 10. Schemat ideowy chromatogra
- Page 102 and 103: Na rys. 13 pokazano kilka chromatog
- Page 104 and 105:
fizycznych i hydrodynamicznych w ko
- Page 106 and 107:
Nie opanowany do końca problem sta
- Page 108 and 109:
Rys. 18. Przykłady reprezentowanyc
- Page 110 and 111:
LITERATURA UZUPEŁNIAJĄCA (pozycje
- Page 112 and 113:
14. Hupe K.P. and Lauer H.H., J. Ch
- Page 114 and 115:
stość izolowanych substancji. Pep
- Page 116 and 117:
(SEC). Ostatnio, do tego celu wykor
- Page 118 and 119:
51243678 90 2 4 6 8 10 12 14 16Rys.
- Page 120 and 121:
jące długie łańcuchy alkilowe,
- Page 122 and 123:
Tabela 2. Modyfikatory fazy ruchome
- Page 124 and 125:
nowymiennych, równorzędnie kation
- Page 126 and 127:
UKŁADY ADSORPCYJNE I ADSORPCYJNO-J
- Page 128 and 129:
CHROMATOGRAFIA ŻELOWAChromatografi
- Page 130 and 131:
CHROMATOGRAFIA ODDZIAŁYWAŃ HYDROF
- Page 132 and 133:
związków. Połączenie elektrofor
- Page 134 and 135:
METODY DETEKCJI W CHROMATOGRAFII PE
- Page 136 and 137:
[25] S.U. Sane, S.M. Cramer, T.M. P