

Electronic Structures - Chemistry
Electronic Structures - Chemistry
Electronic Structures - Chemistry

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
3<br />
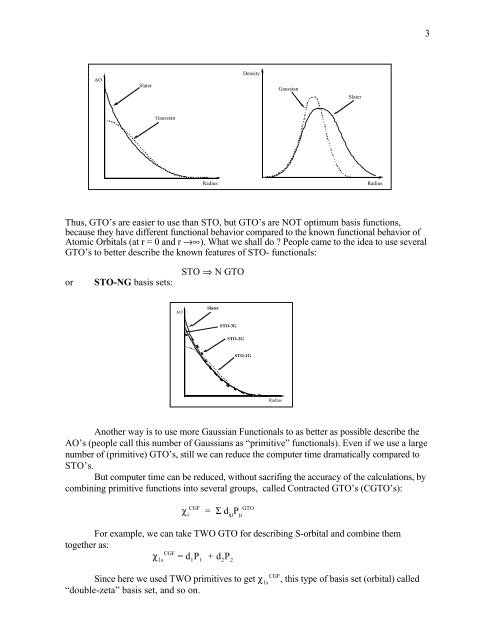
AO<br />
Slater<br />
Density<br />
Gaussian<br />
Slater<br />
Gaussian<br />
Radius<br />
Radius<br />
Thus, GTO’s are easier to use than STO, but GTO’s are NOT optimum basis functions,<br />
because they have different functional behavior compared to the known functional behavior of<br />
Atomic Orbitals (at r = 0 and r →∞). What we shall do People came to the idea to use several<br />
GTO’s to better describe the known features of STO- functionals:<br />
or<br />
STO-NG basis sets:<br />
STO ⇒ N GTO<br />
AO<br />
Slater<br />
STO-3G<br />
STO-2G<br />
STO-1G<br />
Radius<br />
Another way is to use more Gaussian Functionals to as better as possible describe the<br />
AO’s (people call this number of Gaussians as “primitive” functionals). Even if we use a large<br />
number of (primitive) GTO’s, still we can reduce the computer time dramatically compared to<br />
STO’s.<br />
But computer time can be reduced, without sacrifing the accuracy of the calculations, by<br />
combining primitive functions into several groups, called Contracted GTO’s (CGTO’s):<br />
χ i CGF<br />
= Σ d iµ<br />
P µ<br />
GTO<br />
For example, we can take TWO GTO for describing S-orbital and combine them<br />
together as:<br />
χ 1s CGF = d 1<br />
P 1<br />
+ d 2<br />
P 2<br />
Since here we used TWO primitives to get χ 1s CGF , this type of basis set (orbital) called<br />
“double-zeta” basis set, and so on.













