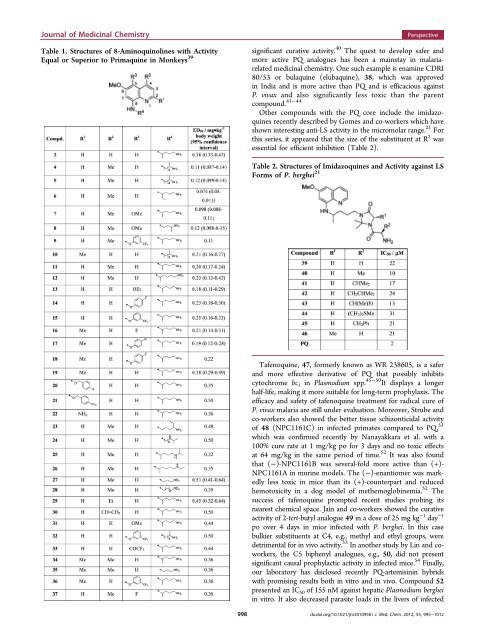
Journal <strong>of</strong> Medicinal ChemistryTable 1. Structures <strong>of</strong> 8-Aminoquinolines with ActivityEqual or Superior to Primaquine in Monkeys 39Perspectivesignificant curative activity. 40 The quest to develop safer andmore active PQ analogues has been a mainstay in malariarelatedmedicinal chemistry. One such example is enamine CDRI80/53 or bulaquine (elubaquine), 38, which was approvedin India and is more active than PQ and is efficacious againstP. vivax and also significantly less toxic than <strong>the</strong> parentcompound. 41−44O<strong>the</strong>r compounds with <strong>the</strong> PQ core include <strong>the</strong> imidazoquinesrecently described by Gomes and co-workers which haveshown interesting anti-LS activity in <strong>the</strong> micromolar range. 21 Forthis series, it appeared that <strong>the</strong> size <strong>of</strong> <strong>the</strong> substituent at R 2 wasessential for efficient inhibition (Table 2).Table 2. Structures <strong>of</strong> Imidazoquines and Activity against LSForms <strong>of</strong> P. berghei 21Tafenoquine, 47, formerly known as WR 238605, is a saferand more effective derivative <strong>of</strong> PQ that possibly inhibitscytochrome bc 1 in Plasmodium spp. 45−50 It displays a longerhalf-life, making it more suitable for long-term prophylaxis. Theefficacy and safety <strong>of</strong> tafenoquine treatment for radical cure <strong>of</strong>P. vivax malaria are still under evaluation. Moreover, Strube andco-workers also showed <strong>the</strong> better tissue schizonticidal activity<strong>of</strong> 48 (NPC1161C) in infected primates compared to PQ, 51which was confirmed recently by Nanayakkara et al. with a100% cure rate at 1 mg/kg po for 3 days and no toxic effectsat 64 mg/kg in <strong>the</strong> same period <strong>of</strong> time. 52 It was also foundthat (−)-NPC1161B was several-fold more active than (+)-NPC1161A in murine models. The (−)-enantiomer was markedlyless toxic in mice than its (+)-counterpart and reducedhemotoxicity in a dog model <strong>of</strong> me<strong>the</strong>moglobinemia. 52 Thesuccess <strong>of</strong> tafenoquine prompted recent studies probing itsnearest chemical space. Jain and co-workers showed <strong>the</strong> curativeactivity <strong>of</strong> 2-tert-butyl analogue 49 in a dose <strong>of</strong> 25 mg kg −1 day −1po over 4 days in mice infected with P. berghei. Inthiscasebulkier substituents at C4, e.g., methyl and ethyl groups, weredetrimental for in vivo activity. 53 In ano<strong>the</strong>r study by Lin and coworkers,<strong>the</strong> C5 biphenyl analogues, e.g., 50, did not presentsignificant causal prophylactic activity in infected mice. 54 Finally,our laboratory has disclosed recently PQ-artemisinin hybridswith promising results both in vitro and in vivo. Compound 52presented an IC 50 <strong>of</strong> 155 nM against hepatic Plasmodium bergheiin vitro. It also decreased parasite loads in <strong>the</strong> livers <strong>of</strong> infected998dx.doi.org/10.1021/jm201095h | J. Med. Chem. 2012, 55, 995−1012
Journal <strong>of</strong> Medicinal ChemistryPerspectiveN-formyl-8-O-methyldioncophylline C, 59, and N-formyl-8-ObenzoyldioncophyllineC, 60, inhibited <strong>the</strong> growth <strong>of</strong> parasiticLS in 98.3% and 52.7%, respectively, at 10 μg/mL. 58 Thesestudies indicated that a dioncophylline A-like biaryl coupling,i.e., with <strong>the</strong> axis between C-7 and C-1′, would favorably influenceactivity. 58mice with a single intraperitoneal (ip) injection <strong>of</strong> 26 μmol/kgdelivered after 3 h <strong>of</strong> infection and nearly abrogated liver infectionafter oral administration. 55Alkaloids. Natural products have been a source for newstarting points for <strong>the</strong> discovery <strong>of</strong> novel drugs againstPlasmodium. 56 Alkaloids, in particular, have been shown topresent interesting anti-LS activity. For example, cambrescidin800, 53, which displays potent blood stage activity, was takenby Diquet and co-workers as a lead for <strong>the</strong> syn<strong>the</strong>sis <strong>of</strong> simpleranalogues with <strong>the</strong> guanidine scaffold. 57 Compound 54 displayedpotent activity (IC 50 = 9.2 μM) against P. yoelii yoeliiLS. However, it did not significantly prolong median survivaltime after a single subcutaneous administration <strong>of</strong> 80 mg/kg inP. berghei-infected mice. 57The alkaloids dioncophylline A, 55, dioncophyllacine A, 56,and ancistrobarterine A, 57, have also displayed excellent anti-LS activity <strong>of</strong> 67.5%, 41.9%, and 51.6% P. berghei growthinhibition, respectively, at 10 μg/mL. In an attempt to elucidate<strong>the</strong> SARs <strong>of</strong> 55 a selection <strong>of</strong> structurally related alkaloidswas examined by François et al. Besides 55 and 57, only 5′-O-demethyl-8-O-methyl-7-epi-dioncophylline A, 58, exertedsignificant inhibitory activity, 48.8% at 10 μg/mL, but wasalso significantly toxic. Also, two dioncophylline C derivatives,O<strong>the</strong>r alkaloids <strong>of</strong> interest include <strong>the</strong> morphinan derivativestazopsine, 61, sinococuline, 62, and 10-epi-tazopsine, 63,which were obtained from <strong>the</strong> bioassay-guided fractionation <strong>of</strong>Strychnopsis thouarsii stem bark extracts. 59 Compounds 61−63exhibited significant and selective inhibitory activity (SI > 13.8)against P. yoelii LS. Tazopsine and sinococulide showed IC 50values <strong>of</strong> 3.1 and 4.5 μM, respectively, while 10-epi-tazopsinewas 5-fold less active and less toxic than 61. Tazopsine displayedan IC 50 <strong>of</strong> 4.2 μM against P. falciparum. 60 Mazier and coworkerssuggested that substitution at C10 was not necessaryfor <strong>the</strong> antimalarial activity. Fur<strong>the</strong>rmore, <strong>the</strong> presence <strong>of</strong> a freehydroxyl group with (R)-stereochemistry enhanced <strong>the</strong> inhibitoryeffect against P. yoelii LS, while <strong>the</strong> presence <strong>of</strong> (S)-stereochemistrysignificantly decreased <strong>the</strong> toxicity on mouse primaryhepatocytes. In contrast, <strong>the</strong> corresponding β-glucoside, 10-epitazosine,64, was inactive, a problem ascribed to <strong>the</strong> steric bulk.Total inhibition was observed for 61 at 7.1 μM, whereas P. yoeliiparasites could still be observed in cultures treated with highconcentrations <strong>of</strong> PQ; 38.6 μM only inhibited 80% <strong>of</strong> parasites. 59Finally, NCP-tazopsine, 65, presented similar activity to 61 butwas significantly less toxic. In this case, oral administration <strong>of</strong>NCP-tazopsine completely protected mice from a sporozoitechallenge and, unlike <strong>the</strong> parent molecule, <strong>the</strong> derivative wasuniquely active against Plasmodium LS. 60999dx.doi.org/10.1021/jm201095h | J. Med. Chem. 2012, 55, 995−1012