Técnicas de análise de DNA e RNA
Técnicas de análise de DNA e RNA
Técnicas de análise de DNA e RNA

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
<strong>Técnicas</strong> <strong>de</strong> <strong>análise</strong> <strong>de</strong> <strong>DNA</strong> e <strong>RNA</strong>
Fundamento e aplicação das técnicas <strong>de</strong> <strong>análise</strong> <strong>de</strong> <strong>DNA</strong><br />
• Extracção, purificação, quantificação e <strong>de</strong>tecção <strong>de</strong> ácidos nucleicos<br />
• Electroforese convencional em gel <strong>de</strong> agarose<br />
• Electroforese em gel <strong>de</strong> campo pulsado (PFGE)<br />
• Hibridação Southern, hibridação dot-blot e hibridação in situ<br />
• Sequenciação <strong>de</strong> <strong>DNA</strong><br />
• Footprinting <strong>de</strong> <strong>DNA</strong><br />
• Retardação em gel<br />
• Mutagénese <strong>de</strong> <strong>DNA</strong>
Extracção, purificação, quantificação e <strong>de</strong>tecção <strong>de</strong> ácidos nucleicos<br />
Extracção e purificação <strong>de</strong> ácidos nucleicos – Utilização <strong>de</strong> Kits comerciais<br />
Quantificação <strong>de</strong> ácidos nucleicos<br />
Método espectrofotométrico (A concentração <strong>de</strong> <strong>DNA</strong> é medida pela absorção a 260 nm, sendo<br />
uma absorvância <strong>de</strong> 1,0 equivalente a 50 µg/ml <strong>de</strong> <strong>DNA</strong> <strong>de</strong> ca<strong>de</strong>ia dupla.)<br />
Método baseado na fluorescência do brometo <strong>de</strong> etídio (Comparação com a fluorescência <strong>de</strong><br />
amostras <strong>de</strong> <strong>DNA</strong> <strong>de</strong> concentração conhecida.)<br />
Detecção <strong>de</strong> ácidos nucleicos<br />
Coloração com brometo <strong>de</strong> etídio<br />
Coloração com nitrato <strong>de</strong> prata<br />
Métodos <strong>de</strong> <strong>de</strong>tecção não radioactivos na hibridação <strong>de</strong> ácidos nucleicos<br />
Marcação das sondas in vitro com:<br />
Corantes fluorescentes ou fluoróforos (fluoresceína e rodamina)<br />
Biotina ou digoxigenina (através <strong>de</strong> ligandos <strong>de</strong> afinida<strong>de</strong> – avidina, estreptavidina ou<br />
anticorpo anti-digoxigenina - acoplados a uma enzima <strong>de</strong> sinalização que converte os<br />
substratos em produtos corados ou quimioluminescentes)<br />
Enzimas (fosfatase alcalina – AP ou peroxidase <strong>de</strong> rábano silvestre – HRP)<br />
Métodos <strong>de</strong> <strong>de</strong>tecção radioactivos na hibridação <strong>de</strong> ácidos nucleicos<br />
Marcação das sondas in vitro com 32 P<br />
Marcação interna (Nick translation, Random primers, PCR)<br />
Marcação terminal 5´ou 3’
Electroforese em gel convencional (1)
Electroforese em gel convencional (2)<br />
A técnica <strong>de</strong> electroforese em gel permite a separação <strong>de</strong> fragmentos <strong>de</strong> <strong>DNA</strong> <strong>de</strong><br />
dimensões diferentes<br />
A matriz <strong>de</strong> gel (ovais laranja) consiste em longas ca<strong>de</strong>ias <strong>de</strong> polímeros, entrelaçadas. A<br />
dimensão das ca<strong>de</strong>ias interligadas, ou poros, <strong>de</strong>pen<strong>de</strong> da concentração <strong>de</strong> agarose ou <strong>de</strong><br />
acrilamida utilizada no gel.<br />
Os poros nos géis <strong>de</strong> agarose são maiores do que nos géis <strong>de</strong> acrilamida. Os primeiros<br />
são utilizados para separar fragmentos gran<strong>de</strong>s <strong>de</strong> <strong>DNA</strong> (≈500 pb a ≈20 kb) e os últimos para<br />
separar fragmentos pequenos <strong>de</strong> <strong>DNA</strong> (1 nucleótido a ≈2 kb).<br />
Sob a acção da corrente eléctrica que passa através do gel os fragmentos são separados,<br />
movendo-se para o pólo positivo a uma taxa inversamente proporcional ao logaritmo da<br />
sua dimensão, formando bandas que são visualizadas por auto-radiografia (fragmentos<br />
radioactivos) ou pela adição <strong>de</strong> um corante fluorescente, como por exemplo o brometo <strong>de</strong><br />
etídio.<br />
Os géis <strong>de</strong> agarose são horizontais, mas os <strong>de</strong> acrilamida são verticais (preparados entre<br />
duas placas <strong>de</strong> vidro com um afastamento <strong>de</strong> alguns milímetros apenas) porque o oxigénio do<br />
ar inibe a polimerização da acrilamida.
Electroforese em gel <strong>de</strong> campo pulsado (PFGE) (1)<br />
A técnica <strong>de</strong> electroforese em gel <strong>de</strong> campo pulsado – PFGE é utilizada para separar cromossomas<br />
diferentes, como por exemplo <strong>de</strong> Saccharomyces cerevisiae, cujas dimensões variam entre 220 000 a<br />
2,5 milhões <strong>de</strong> pares <strong>de</strong> nucleótidos, ou moléculas <strong>de</strong> <strong>DNA</strong> com 10 7 pares <strong>de</strong> nucleótidos. O <strong>DNA</strong> é<br />
corado com brometo <strong>de</strong> etídio.
Electroforese em gel <strong>de</strong> campo pulsado (PFGE) (2)<br />
Definição das condições: Nº <strong>de</strong> fases; duração dos pulsos; duração <strong>de</strong> cada fase; amperagem.<br />
Exemplo: Fase 1; pulso 1 segundo; duração da fase 36 minutos; 180 mA<br />
Fase 2; pulso 2 segundos; duração da fase 36 minutos; 180 mA
Hibridação Southern e hibridação Northern (1)<br />
As técnicas <strong>de</strong> hibridação Southern e hibridação Northern<br />
permitem, respectivamente, a <strong>de</strong>tecção <strong>de</strong> <strong>DNA</strong>s ou <strong>de</strong> <strong>RNA</strong>s<br />
específicos por hibridação com sondas apropriadas, <strong>de</strong> <strong>DNA</strong> e<br />
<strong>RNA</strong>.<br />
Neste exemplo, a sonda <strong>de</strong> <strong>DNA</strong> é <strong>de</strong>tectada pela sua<br />
radioactivida<strong>de</strong>, mas as sondas também po<strong>de</strong>m ser <strong>de</strong>tectadas por<br />
métodos não radioactivos.
Hibridação Southern e hibridação Northern (2)<br />
Estas técnicas envolvem as seguintes etapas:<br />
(A) Misturas <strong>de</strong> moléculas <strong>de</strong> <strong>RNA</strong> <strong>de</strong> ca<strong>de</strong>ia simples (Hibridação Northern) ou <strong>de</strong> moléculas<br />
<strong>de</strong> <strong>DNA</strong> <strong>de</strong> ca<strong>de</strong>ia dupla obtidas por digestão com enzimas <strong>de</strong> restrição (Hibridação Southern)<br />
são separadas por electroforese com base na dimensão.<br />
(B) As moléculas <strong>de</strong> <strong>RNA</strong> ou os fragmentos <strong>de</strong> <strong>DNA</strong> <strong>de</strong>snaturados por exposição do <strong>DNA</strong> a<br />
condições alcalinas <strong>de</strong>snaturantes, são transferidos para membranas <strong>de</strong> nitrocelulose ou <strong>de</strong><br />
nylon.<br />
(C) A membrana, contendo os ácidos nucleicos ligados, é cuidadosamente removida do gel.<br />
(D) A membrana é <strong>de</strong>pois colocada num saco <strong>de</strong> plástico selado ou numa garrafa <strong>de</strong> hibridação,<br />
juntamente com a solução <strong>de</strong> hibridação (uma solução salina tamponada) e a sonda <strong>de</strong> <strong>DNA</strong> ou<br />
<strong>de</strong> <strong>RNA</strong>, radioactivamente marcada e na forma <strong>de</strong> ca<strong>de</strong>ia simples, por um período <strong>de</strong> tempo<br />
prolongado em condições que favorecem a hibridação.<br />
(E) A membrana é em seguida removida do saco ou da garrafa <strong>de</strong> hibridação e lavada<br />
vigorosamente, <strong>de</strong> modo que apenas as moléculas <strong>de</strong> sonda que hibridaram com o <strong>RNA</strong> ou o<br />
<strong>DNA</strong> imobilizado na membrana permanecem ligadas. Após auto-radiografia, o <strong>DNA</strong> ou o<br />
<strong>RNA</strong> que hibridou com a sonda marcada é visualizado como uma banda.
Hibridação dot-blot
Estratégias <strong>de</strong> sequenciação shotgun do genoma humano<br />
A sequenciação shotgun significa que o <strong>DNA</strong> genómico original é aleatoriamente clivado em pequenos fragmentos.<br />
Estratégia da empresa pública<br />
Digestão parcial<br />
Clonagem em vectores BAC<br />
Sonicação e reparação das<br />
extremida<strong>de</strong>s dos fragmentos<br />
Clonagem<br />
Vector M13 ou<br />
Fagemídio<br />
Estratégia da empresa privada<br />
Celera
Sequenciação <strong>de</strong> insertos gran<strong>de</strong>s
Pesquisa <strong>de</strong> regiões codificantes numa sequência <strong>de</strong> <strong>DNA</strong> (1)
Pesquisa <strong>de</strong> regiões codificantes numa sequência <strong>de</strong> <strong>DNA</strong> (2)<br />
(A) Qualquer região da sequência <strong>de</strong> <strong>DNA</strong>, em princípio, codifica seis sequências diferentes <strong>de</strong><br />
aminoácidos, porque as três grelhas diferentes <strong>de</strong> leitura são utilizadas para interpretar a sequência<br />
<strong>de</strong> nucleótidos em cada ca<strong>de</strong>ia.<br />
A sequência <strong>de</strong> nucleótidos é sempre lida na direcção 5’→3’ da ca<strong>de</strong>ia e codifica um<br />
polipéptido a partir da extremida<strong>de</strong> amínica (N) para a extremida<strong>de</strong> carboxílica (C).<br />
Na leitura <strong>de</strong> uma sequência nucleotídica aleatória numa grelha particular é encontrado, em<br />
média, um sinal <strong>de</strong> paragem da síntese proteica em 21 aminoácidos (um em 63 nucleótidos).<br />
Na sequência <strong>de</strong> 48 pb, os codões stop estão representados a ver<strong>de</strong>, e apenas a grelha <strong>de</strong> leitura<br />
2 não tem codão stop.<br />
(B) Pesquisa na sequência <strong>de</strong> <strong>DNA</strong> <strong>de</strong> 1700 pb <strong>de</strong> uma possível sequência codificante <strong>de</strong> uma<br />
proteína. A informação está apresentada como em (A), com cada codão stop representado a ver<strong>de</strong>.<br />
Todas as regiões entre possíveis codões <strong>de</strong> iniciação e terminação da síntese proteica estão<br />
representadas como barras vermelhas. Apenas a grelha <strong>de</strong> leitura 1 codifica realmente uma proteína,<br />
<strong>de</strong> 475 aminoácidos.
Aplicações da sequenciação <strong>de</strong> genomas
Técnica <strong>de</strong> footprinting <strong>de</strong> <strong>DNA</strong> (1)<br />
Esta técnica permite <strong>de</strong>tectar interacções <strong>DNA</strong>-proteína<br />
Figure 8-54. © 2002 by Bruce Alberts, Alexan<strong>de</strong>r Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter.
Técnica <strong>de</strong> footprinting <strong>de</strong> <strong>DNA</strong> (2)<br />
(A) A técnica <strong>de</strong> footprinting <strong>de</strong> <strong>DNA</strong> requer uma molécula <strong>de</strong> <strong>DNA</strong> previamente marcada numa<br />
das extremida<strong>de</strong>s.<br />
A proteína representada na figura tem uma ligação forte com sete nucleótidos da sequência <strong>de</strong><br />
<strong>DNA</strong> específica, protegendo esses nucleótidos do agente <strong>de</strong> clivagem, a enzima DNase I.<br />
Na reacção paralela <strong>de</strong> clivagem com DNase I realizada sem a proteína <strong>de</strong> ligação a <strong>DNA</strong>,<br />
é visualizado no gel <strong>de</strong> acrilamida <strong>de</strong>snaturante o conjunto completo <strong>de</strong> bandas (não está<br />
representado).<br />
Os resultados são analisados, em paralelo, com as quatro reacções <strong>de</strong> sequenciação pelo<br />
método <strong>de</strong> Sanger do fragmento em estudo, o que permite <strong>de</strong>terminar a sequência <strong>de</strong> <strong>DNA</strong><br />
reconhecida pela proteína. A i<strong>de</strong>ntificação dos nucleótidos envolvidos na interacção com a<br />
proteina é obtida por mutagénese in vitro.<br />
(B) O footprint foi realizado para <strong>de</strong>terminar o local <strong>de</strong> ligação <strong>de</strong> uma proteína humana que<br />
estimula a transcrição <strong>de</strong> genes eucarióticos específicos. Estes resultados indicaram que o local <strong>de</strong><br />
ligação se situa a cerca <strong>de</strong> 60 nucleótidos a montante do local <strong>de</strong> início da transcrição.
Retardação em gel (1)<br />
Esta técnica permite <strong>de</strong>tectar interacções <strong>DNA</strong>-proteína
Retardação em gel (2)<br />
Este método permite <strong>de</strong>terminar, após sequenciação, a sequência <strong>de</strong> <strong>DNA</strong> reconhecida por<br />
uma proteína reguladora <strong>de</strong> um gene. A i<strong>de</strong>ntificação dos nucleótidos envolvidos na<br />
interacção com a proteína é obtida por mutagénese in vitro.<br />
A proteína relevante é purificada e misturada com milhões <strong>de</strong> diferentes fragmentos curtos <strong>de</strong><br />
<strong>DNA</strong>, cada um com uma sequência diferente <strong>de</strong> nucleótidos. Um conjunto <strong>de</strong>stes fragmentos po<strong>de</strong><br />
ser produzido programando um sintetizador <strong>de</strong> <strong>DNA</strong>, aparelho que sintetiza quimicamente <strong>DNA</strong><br />
com a sequência pretendida. Por exemplo, há 4 11 , ou aproximadamente 42 milhões <strong>de</strong> sequências<br />
possíveis para um fragmento <strong>de</strong> <strong>DNA</strong> <strong>de</strong> 11 nucleótidos.<br />
Os fragmentos <strong>de</strong> <strong>DNA</strong> <strong>de</strong> ca<strong>de</strong>ia dupla que se ligam fortemente à proteína reguladora do gene<br />
são <strong>de</strong>pois separados dos fragmentos <strong>de</strong> <strong>DNA</strong> que não se ligam por retardação em gel.<br />
Após separação dos complexos proteína-<strong>DNA</strong> do <strong>DNA</strong> livre, os fragmentos <strong>de</strong> <strong>DNA</strong> são<br />
removidos da proteína, e são realizados vários ciclos adicionais do mesmo processo <strong>de</strong> selecção. As<br />
sequências <strong>de</strong> nucleótidos dos fragmentos <strong>de</strong> <strong>DNA</strong> que permanecem através <strong>de</strong> múltiplos ciclos <strong>de</strong><br />
selecção são <strong>de</strong>terminadas, e uma sequência consenso <strong>de</strong> <strong>DNA</strong> reconhecida pela proteína po<strong>de</strong> ser<br />
obtida.
Retardação em gel (3)<br />
Esta técnica tem diferentes <strong>de</strong>signações: Electroforetic mobility shift assay (EMSA); Gelshift; Gel<br />
retardation; Bandshift assay.<br />
Figure 7-29. © 2002 by Bruce Alberts, Alexan<strong>de</strong>r Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter.
Retardação em gel (4)<br />
O fundamento da técnica <strong>de</strong> retardação em gel está esquematizado em (A).<br />
Neste exemplo, o extracto proteico <strong>de</strong> uma linha celular produtora <strong>de</strong> anticorpo é misturado com o<br />
fragmento <strong>de</strong> <strong>DNA</strong> radioactivo contendo cerca <strong>de</strong> 160 nucleótidos da sequência <strong>de</strong> <strong>DNA</strong> reguladora<br />
do gene que codifica a ca<strong>de</strong>ia leve do anticorpo produzido pela linha celular.<br />
O efeito das proteínas do extracto na mobilida<strong>de</strong> do fragmento <strong>de</strong> <strong>DNA</strong> é analisado por<br />
electroforese em gel <strong>de</strong> poliacrilamida, seguida <strong>de</strong> auto-radiografia, comparando com a mobilida<strong>de</strong> do<br />
fragmento <strong>de</strong> <strong>DNA</strong> sem adição do extracto.<br />
Os fragmentos <strong>de</strong> <strong>DNA</strong> livres migram rapidamente para o fundo do gel, enquanto os<br />
fragmentos ligados a proteínas são retardados; as seis bandas retardadas sugerem que o extracto<br />
contém seis proteínas diferentes <strong>de</strong> ligação a sequências específicas <strong>de</strong> <strong>DNA</strong> (C1 a C6), que se ligam<br />
a este fragmento. (Para simplificar, os fragmentos <strong>de</strong> <strong>DNA</strong> com mais <strong>de</strong> uma proteína ligada foram<br />
omitidos da figura).<br />
Em (B), o extracto foi fraccionado por uma técnica padrão <strong>de</strong> cromatografia (em cima), e cada<br />
fracção foi misturada com o fragmento <strong>de</strong> <strong>DNA</strong> radioactivo, aplicada num poço do gel <strong>de</strong><br />
poliacrilamida e analisada como em (A).
Purificação <strong>de</strong> proteínas <strong>de</strong> ligação a <strong>DNA</strong> por cromatografia <strong>de</strong> afinida<strong>de</strong> (1)<br />
Figure 7-30. © 2002 by Bruce Alberts, Alexan<strong>de</strong>r Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter.
Purificação <strong>de</strong> proteínas <strong>de</strong> ligação a <strong>DNA</strong> por cromatografia <strong>de</strong> afinida<strong>de</strong> (2)<br />
Na etapa 1, as proteínas <strong>de</strong> ligação a <strong>DNA</strong> são separadas das restantes proteínas celulares numa coluna<br />
com diferentes sequências <strong>de</strong> <strong>DNA</strong>.<br />
A fraca afinida<strong>de</strong> por <strong>DNA</strong> não específico resulta <strong>de</strong> atracções iónicas, sendo as proteínas removidas<br />
por uma solução com uma concentração mo<strong>de</strong>rada <strong>de</strong> sal.<br />
Na etapa 2, a coluna contém apenas uma sequência particular <strong>de</strong> <strong>DNA</strong>, e as proteínas retidas <strong>de</strong>vido a<br />
interacções não específicas são eluídas por soluções <strong>de</strong> concentração mo<strong>de</strong>rada <strong>de</strong> sal, <strong>de</strong>ixando na<br />
coluna as proteínas (em geral uma) fortemente ligadas à sequência <strong>de</strong> <strong>DNA</strong>. Estas proteínas são eluídas<br />
da coluna por soluções contendo uma concentração muito elevada <strong>de</strong> sal.<br />
Um método alternativo <strong>de</strong> purificação <strong>de</strong> uma proteína <strong>de</strong> ligação a <strong>DNA</strong> consiste na triagem <strong>de</strong><br />
uma biblioteca <strong>de</strong> c<strong>DNA</strong> com uma sonda <strong>de</strong> <strong>DNA</strong> que contém o local <strong>de</strong> ligação à proteína.
Mutagénese sítio-específica com primer mutagénico<br />
Apesar da falta <strong>de</strong> complementarida<strong>de</strong> dos nucleótidos internos, o emparelhamento do primer<br />
mutagénico é possível, sendo a segunda ca<strong>de</strong>ia sintetizada pela polimerase <strong>de</strong> <strong>DNA</strong>, e as suas<br />
extremida<strong>de</strong>s ligadas pela ligase <strong>de</strong> <strong>DNA</strong>.<br />
Os homodúplices são i<strong>de</strong>ntificados por hibridação molecular, utilizando o primer mutagénico como sonda<br />
oligonucleotídica alelo-específica, ou por PCR alelo-específico.
Mutagénese sítio-dirigida por PCR<br />
• Deve ser utilizada uma<br />
polimerase termoestável<br />
com capacida<strong>de</strong> <strong>de</strong><br />
revisão <strong>de</strong> provas para<br />
minimizar eventuais erros.<br />
• O <strong>DNA</strong> mol<strong>de</strong>, isolado<br />
<strong>de</strong> estirpes dam + , é<br />
metilado e susceptível à<br />
digestão com DpnI, uma<br />
endonuclease específica<br />
<strong>de</strong> <strong>DNA</strong> metilado e<br />
hemimetilado.
Fundamento e aplicação das técnicas <strong>de</strong> <strong>análise</strong> <strong>de</strong> <strong>RNA</strong><br />
• Hibridação Northern<br />
• Mapeamento <strong>de</strong> extremida<strong>de</strong>s 5’ e 3’ do m<strong>RNA</strong> com nuclease S1<br />
• Mapeamento <strong>de</strong> extremida<strong>de</strong>s 5’ do m<strong>RNA</strong> por extensão do primer<br />
• Microarrays <strong>de</strong> <strong>DNA</strong>
Hibridação Northern<br />
Para além da hibridação Northern, as técnicas <strong>de</strong> RT-PCR e<br />
hibridação in situ permitem analisar em que tecidos ou<br />
condições experimentais os genes se expressam.<br />
A hibridação Northern permite:<br />
• Detectar a presença <strong>de</strong> m<strong>RNA</strong><br />
específico<br />
• Estimar a dimensão do m<strong>RNA</strong>,<br />
relativamente a <strong>RNA</strong>s marcadores<br />
• Estimar a quantida<strong>de</strong> relativa do<br />
m<strong>RNA</strong> (por comparação do sinal<br />
<strong>de</strong> hibridação com o <strong>de</strong> outros<br />
genes que são expressos em níveis<br />
conhecidos).<br />
• Detectar a presença <strong>de</strong> transcritos<br />
diferentes <strong>de</strong> um gene específico<br />
(splicing alternativo, promotores<br />
alternativos).
Mapeamento <strong>de</strong> extremida<strong>de</strong>s 5’ do m<strong>RNA</strong><br />
Técnica <strong>de</strong> protecção da nuclease S1 Técnica <strong>de</strong> extensão do primer<br />
A extremida<strong>de</strong> 5’ do m<strong>RNA</strong> correspon<strong>de</strong> ao local <strong>de</strong> início da transcrição.
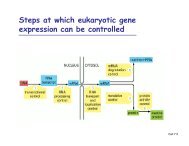
Análise da expressão génica com microarrays <strong>de</strong> <strong>DNA</strong> (1)<br />
Cada spot contém 10 6 -10 9 cópias da mesma<br />
sequência <strong>de</strong> c<strong>DNA</strong> <strong>de</strong> várias centenas <strong>de</strong><br />
pares <strong>de</strong> bases.<br />
c<br />
Perfect match<br />
Mismatch<br />
Cada gene está representado por 20 pares <strong>de</strong><br />
oligonucleótidos (PM e MM) diferentes <strong>de</strong><br />
ca<strong>de</strong>ia simples com 20-25 nt.
Análise da expressão génica com microarrays <strong>de</strong> <strong>DNA</strong> (2)<br />
c<strong>DNA</strong> marcado<br />
Nível <strong>de</strong> expressão<br />
X - elevado em 1<br />
Y – elevado em 2<br />
Transcritase reversa<br />
c<strong>DNA</strong><br />
Z – idêntico em 1 e 2<br />
c<strong>DNA</strong> marcado<br />
Avidina conjugada<br />
com um fluoróforo
Análise da expressão génica com microarrays <strong>de</strong> <strong>DNA</strong> (3)<br />
Os oligos MM contêm uma única base mismatch e servem para controlar a hibridação inespecífica.<br />
O controlo positivo é o gene da actina, gene expresso constitutivamente.<br />
O controlo negativo incluído nos microarrays <strong>de</strong> c<strong>DNA</strong> serve para normalizar o background ou<br />
hibridação não específica.<br />
Para <strong>de</strong>terminar o sinal <strong>de</strong> um gene particular, os sinais dos 20 oligos PM são somados e os sinais<br />
dos 20 oligos MM são subtraídos do total.<br />
Em (C) um dos nucleótidos está conjugado com um fluoróforo.<br />
X – representa genes hipotéticos presentes em níveis elevados na amostra 1; Y – níveis elevados na<br />
amostra 2; Z – níveis idênticos nas amostras 1 e 2.<br />
Para este tipo <strong>de</strong> <strong>análise</strong> da expressão baseada na hibridação multiplex muitas empresas<br />
comercializam (A), mas (B) é comercializado como GeneChips apenas pela Companhia <strong>de</strong><br />
Biotecnologia US Affymetrix Inc..