Adrenogenitales Syndrom (AGS) OMIM 201910 mit 21 ... - IMD-Lab
Adrenogenitales Syndrom (AGS) OMIM 201910 mit 21 ... - IMD-Lab
Adrenogenitales Syndrom (AGS) OMIM 201910 mit 21 ... - IMD-Lab

Sie wollen auch ein ePaper? Erhöhen Sie die Reichweite Ihrer Titel.
YUMPU macht aus Druck-PDFs automatisch weboptimierte ePaper, die Google liebt.
<strong>Adrenogenitales</strong> <strong>Syndrom</strong> (<strong>AGS</strong>) <strong>OMIM</strong> <strong>201910</strong><br />
<strong>mit</strong> <strong>21</strong>-Hydroxylase Mangel<br />
1. Bedeutung<br />
Das Adrenogenitale <strong>Syndrom</strong> (<strong>AGS</strong>) ist eine autosomal-rezessiv vererbte Krankheit,<br />
die durch einen Defekt eines an der Biosynthese von Cortisol beteiligten Enzyms<br />
hervorgerufen wird (1). In der kaukasischen Bevölkerung ist 1:10'000 Personen von<br />
der schweren klassischen Form und 1:1000 von der milden nicht-klassischen Form des<br />
<strong>AGS</strong> betroffen. Bei Italienern, Yugoslawen, Ashkenazischen Juden und Lateinamerikanern<br />
kommt die nicht-klassische Form des <strong>AGS</strong> sogar noch häufiger vor (2, 3).<br />
Die Ursache ist in 95% der Fälle ein Defekt der <strong>21</strong>-Hydroxylase, welche von dem Gen<br />
CYP<strong>21</strong>A2 auf dem Chromosom 6p<strong>21</strong>.3 codiert wird (4).<br />
Aufgrund der fehlenden oder herabgesetzten Enzymaktivität der <strong>21</strong>-Hydroxylase fällt<br />
auf der einen Seite vermehrt 17-Hydroxyprogesteron an, auf der anderen Seite kommt<br />
es zu einer herabgesetzten Synthese von Aldosteron und Cortisol. Durch den<br />
vermehrten Anfall von Metaboliten der Steroidsynthese und Ausbleiben der<br />
Rückkopplung durch Cortisol kommt es zu einer vermehrten Ausschüttung von ACTH<br />
und sekundär zur Vergrösserung der Nebennierenrinde sowie zu einem Anstieg von<br />
Testosteron (5, 6).<br />
Der Enzymmangel kann sich - in Abhängigkeit von der zugrundeliegenden Mutation -<br />
in verschiedenen Formen manifestieren. Die Mutationen lassen sich nach Schweregrad<br />
der Symptome in 4 Klassen unterteilen: In der Gruppe SW (salt-wasting) führt die<br />
Mutation zur kompletten Inaktivierung der <strong>21</strong>-Hydroxylase Enzymaktivität. Es kommt<br />
bereits in utero zur Virilisierung der weiblichen Feten und nach der Geburt zu einem<br />
lebensbedrohenden Salzverlust. Zu dieser Gruppe gehören die vom Pseudogen<br />
herkommenden Nullmutationen: 8bp Deletion im Exon 3, Mutationscluster im Exon 6,<br />
L307Frameshift, Q318Stop, R356W. In der Gruppe SV (simple virilizing) führt die<br />
Mutation ebenfalls zu einer pränatalen Virilisierung der Genitalien und gelegentlich zu<br />
leichten, jedoch nicht lebensbedrohenden Symptomen eines Salzverlusts. Ein Beispiel<br />
dieser Gruppe ist die Mutation I172N. Die Gruppe NC (non-classic) trägt Mutationen,<br />
die deutlich mildere Verläufe zeigen. Die ersten Symptome treten erst in der Pubertät<br />
auf. Bei weiblichen Jugendlichen kommt es zu prämaturer Pubarche (vorzeitige<br />
Entwicklung der Schambehaarung), Hirsutismus, Zyklus- und Fertilitätsstörungen,<br />
tiefer Stimmlage und Akne. Männliche fallen höchstens durch eine Pubertas praecox<br />
auf. Die Mutationen V281L und P30L gehören zu dieser Gruppe. Die letzte Gruppe wird<br />
als "normal" bezeichnet, weil die Mutationen keine Änderung des Phänotyps<br />
hervorrufen. Bis anhin veröffentlichte Normalvarianten sind 9_10insL, K102R, D183E,<br />
S268T und N493S (5, 6).<br />
Die Möglichkeit, den Schweregrad der Krankheit aufgrund der Mutation vorherzusagen,<br />
hat direkte Auswirkungen auf die Behandlung von <strong>AGS</strong> Patienten. So wurde schon<br />
1984 die pränatale Behandlung eines <strong>AGS</strong> Falls beschrieben, bei der die Virilisierung<br />
durch frühe Gabe von plazentagängigen Glucocorticoiden weitgehend verhindert<br />
werden konnte (7).<br />
________________________________________________________________________________________________________________________<br />
12.04.2012 <strong>Adrenogenitales</strong> <strong>Syndrom</strong> (<strong>AGS</strong>) <strong>IMD</strong> Institut für medizinische & molekulare Diagnostik AG, Zürich
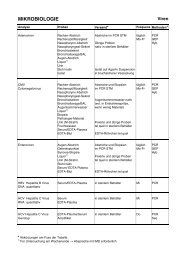
2. Nachweismethoden<br />
3. Indikationen<br />
Zur molekularen Abklärung des <strong>21</strong>-Hydroxylase Mangels führen wir eine MLPA<br />
Analyse (8) und die Sequenzierung des CYP<strong>21</strong>A2 Gens durch (9, 10, 11). Das<br />
CYP<strong>21</strong>A2 Gen liegt 30kb entfernt vom nicht-funktionellen Pseudogen CYP<strong>21</strong>A2P auf<br />
Chromosom 6. Beide Gene sind etwa 3kb lang und über 95% homolog. Deletionen,<br />
Konversionen und die häufigsten Punktmutationen im CYP<strong>21</strong>A2 Gen rühren von<br />
Rearrangements zwischen dem funktionellen und dem Pseudogen her. Die zehn<br />
häufigsten Punktmutationen werden so vom Pseudogen auf das CYP<strong>21</strong>A2 Gen<br />
übertragen und beeinflussen die Enzymaktivität. Mittels MLPA werden grosse<br />
Deletionen/Duplikationen des CYP<strong>21</strong>A2 Gens erfasst, welche mehr als 20% der Fälle<br />
von <strong>21</strong>-Hydroxylase Mangel ausmachen. Zur Sequenzierung werden vier spezifische<br />
Fragmente von CYP<strong>21</strong>A2 - und nicht des Pseudogens - amplifiziert und die kodierende<br />
Region der Exons 1-10 inklusive Introns sowie 350 Nukleotide im 5'- und 500<br />
Nukleotide im 3'-Bereich sequenziert. Mit dieser Methode werden Punktmutationen<br />
sowie kleine Deletionen und Insertionen erfasst. Durch die Kombination von<br />
Sequenzierung und MLPA Analyse werden 98% der krankheitsverursachenden Allele<br />
von Betroffenen und Anlageträgern erfasst.<br />
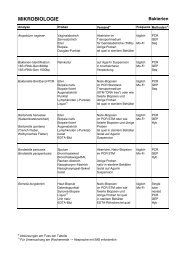
• Neugeborene <strong>mit</strong> Virilisierung, intersexuellem Genitale oder Salzverlust<br />
• Guthrie Test <strong>mit</strong> erhöhtem 17-OH-Progesteron<br />
• Pränataldiagnostik<br />
• Geschwister und Partner von <strong>AGS</strong>-Indexpatienten<br />
• Frauen <strong>mit</strong> Hyperandrogenämie und Fertilitätsstörungen nach endokrinologischer<br />
Differentialdiagnostik<br />
4. Untersuchungsmaterialien<br />
• 2-5 ml EDTA Blut<br />
• Chorionzottenbiopsie, Fruchtwasser<br />
______________________________________________________________________________________________<br />
Literatur:<br />
[1] New MI, Dupont B, Grumbach K and Levine LS in The Metabolic Basis of Inherited Disease (eds Stanbury JB,<br />
Wyngaarden JB, Fredrickson DS, Goldstein JL and Brown MS) McGraw-Hill, New York, 1982: 973−1000.<br />
[2] Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, and New MI. High frequency of nonclassical steroid<br />
<strong>21</strong>-hydroxylase deficiency. Am J Hum Genet. 1985; 37: 650–667.<br />
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1684620/<br />
[3] van der Kamp HJ and Wit JM. Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol. 2004;<br />
151 Suppl 3:U71-5. http://eje-online.org/content/151/Suppl_3/U71.long<br />
[4] White PC, New MI and Dupont B. Structure of human steroid <strong>21</strong>-hydroxylase genes. Proc. Natl. Acad. Sci. USA<br />
1986: 83, 5111-5115. http://www.pnas.org/content/83/14/5111.long<br />
[5] Wedell A. Molecular genetics of congenital adrenal hyperplasia (<strong>21</strong>-hydroxylase deficiency): implications for<br />
diagnosis, prognosis and treatment. Acta Paediatr. 1998; 87: 159-64.<br />
[6] White PC and Speiser PW. Congenital Adrenal Hyperplasia due to <strong>21</strong>-Hydroxylase Deficiency.<br />
Endocrine Reviews 2000; <strong>21</strong>: 245-291. http://edrv.endojournals.org/content/<strong>21</strong>/3/245.full.pdf+html<br />
[7] David M and Forest MG. Prenatal treatment of congenital adrenal hyperplasia resulting from <strong>21</strong>-hydroxylase<br />
deficiency. J Pediatr. 1984; 105: 799-803.<br />
[8[ http://www.mlpa.com, SALSA MLPA P050 CAH<br />
[9] Wedell A., Ritzén EM, Haglund-Stengler B and Luthman H. Steroid <strong>21</strong>-hydroxylase deficiency: Three additional<br />
mutated alleles and establishment of phenotype genotype relationships of common mutations. Proc. Natl. Acad.<br />
Sci. USA 1992; 89: 7232-7236. http://www.pnas.org/content/89/15/7232.long<br />
________________________________________________________________________________________________________________________<br />
12.04.2012 <strong>Adrenogenitales</strong> <strong>Syndrom</strong> (<strong>AGS</strong>) <strong>IMD</strong> Institut für medizinische & molekulare Diagnostik AG, Zürich
[10] Day DJ, Speiser PW, White PC and Barany F. Detection of steroid <strong>21</strong>-hydroxylase alleles using gene-specific<br />
PCR and a multiplexed ligation detection reaction. Genomics 1995; 29: 152-62.<br />
[11] Blanché H, Vexiau P, Clauin S, Le Gall I, Fiet J, Mornet E, Dausset J and Bellanné-Chantelot C. Exhaustive<br />
screening of the <strong>21</strong>-hydroxylase gene in a population of hyperandrogenic women. Hum Genet. 1997; 101: 56-60.<br />
________________________________________________________________________________________________________________________<br />
12.04.2012 <strong>Adrenogenitales</strong> <strong>Syndrom</strong> (<strong>AGS</strong>) <strong>IMD</strong> Institut für medizinische & molekulare Diagnostik AG, Zürich