

Coat Variation in the Domestic Dog is Governed - Biology @ Davidson
Coat Variation in the Domestic Dog is Governed - Biology @ Davidson
Coat Variation in the Domestic Dog is Governed - Biology @ Davidson

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
nucleotide polymorph<strong>is</strong>m (SNP) data sets us<strong>in</strong>g<br />
<strong>the</strong> Affymetrix version 2.0 can<strong>in</strong>e SNP chip (8, 9).<br />
The first data set cons<strong>is</strong>ted of 96 dachshunds segregat<strong>in</strong>g<br />
three coat varieties: wire-haired with<br />
furn<strong>is</strong>h<strong>in</strong>gs, smooth, and long-haired without furn<strong>is</strong>h<strong>in</strong>gs.<br />
The second data set compr<strong>is</strong>ed 76 Portuguese<br />
water dogs (PWDs), segregat<strong>in</strong>g <strong>the</strong> curl<br />
phenotype. The f<strong>in</strong>al data set, termed CanMap,<br />
<strong>in</strong>cluded 903 dogs from 80 breeds represent<strong>in</strong>g a<br />
wide variety of phenotypes. An additional data<br />
set used to map furn<strong>is</strong>h<strong>in</strong>gs <strong>in</strong>cluded a panel of<br />
microsatellite markers (10), genotyped on a 96dachshund<br />
pedigree segregat<strong>in</strong>g all three coat<br />
varieties.<br />
The same strategy was used to map all three<br />
traits. First, a genome-wide association study<br />
(GWAS) with<strong>in</strong> a breed segregat<strong>in</strong>g <strong>the</strong> phenotype<br />
was conducted to determ<strong>in</strong>e <strong>the</strong> most strongly<br />
associated locus. To rule out false-positives<br />
caused by population structure with<strong>in</strong> <strong>the</strong> breeds<br />
(11), we did a second GWAS that used <strong>the</strong><br />
CanMap data set divided <strong>in</strong>to cases and controls<br />
A<br />
B<br />
-log 10 (pval)<br />
C<br />
-log 10 (pval)<br />
D<br />
25<br />
20<br />
15<br />
10<br />
5<br />
250<br />
200<br />
150<br />
100<br />
50<br />
1<br />
1<br />
based on <strong>the</strong> presence or absence of <strong>the</strong> phenotype<br />
<strong>in</strong> question. F<strong>in</strong>e-mapp<strong>in</strong>g of significant,<br />
concordant peaks was used to def<strong>in</strong>e <strong>the</strong> smallest<br />
shared haplotype, followed by sequenc<strong>in</strong>g to identify<br />
<strong>the</strong> putative causative mutations. Each mutation<br />
was validated <strong>in</strong> a large panel of at least<br />
661 dogs from 108 breeds, <strong>in</strong>clud<strong>in</strong>g cases and<br />
controls for all phenotypes (table S1).<br />
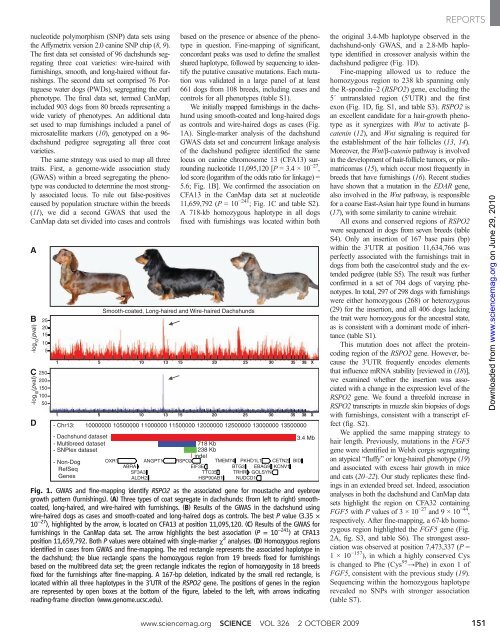
We <strong>in</strong>itially mapped furn<strong>is</strong>h<strong>in</strong>gs <strong>in</strong> <strong>the</strong> dachshund<br />
us<strong>in</strong>g smooth-coated and long-haired dogs<br />
as controls and wire-haired dogs as cases (Fig.<br />
1A). S<strong>in</strong>gle-marker analys<strong>is</strong> of <strong>the</strong> dachshund<br />
GWAS data set and concurrent l<strong>in</strong>kage analys<strong>is</strong><br />
of <strong>the</strong> dachshund pedigree identified <strong>the</strong> same<br />
locus on can<strong>in</strong>e chromosome 13 (CFA13) surround<strong>in</strong>g<br />
nucleotide 11,095,120 [P =3.4×10 −27 ,<br />
lod score (logarithm of <strong>the</strong> odds ratio for l<strong>in</strong>kage) =<br />
5.6; Fig. 1B]. We confirmed <strong>the</strong> association on<br />
CFA13 <strong>in</strong> <strong>the</strong> CanMap data set at nucleotide<br />
11,659,792 (P =10 −241 ; Fig. 1C and table S2).<br />
A 718-kb homozygous haplotype <strong>in</strong> all dogs<br />
fixed with furn<strong>is</strong>h<strong>in</strong>gs was located with<strong>in</strong> both<br />
- Chr13: 10000000 10500000 11000000 11500000 12000000 12500000 13000000 13500000<br />
- Dachshund dataset<br />
- Multibreed dataset<br />
- SNPlex dataset<br />
- Non-<strong>Dog</strong><br />
RefSeq<br />
Genes<br />
5<br />
5<br />
Smooth-coated, Long-haired and Wire-haired Dachshunds<br />
OXR1<br />
10 13 15<br />
20<br />
25 30 3538 X<br />
10 13 15<br />
20<br />
25 30 3538 X<br />
ANGPT1<br />
ABRA<br />
SF3A3<br />
ALDH2<br />
718 Kb<br />
238 Kb<br />
<strong>in</strong>del<br />
RSPO2<br />
EIF3E<br />
TTC35<br />
HSP90AB1<br />
TMEM74<br />
PKHD1L1 CETN2 BID<br />
BTG3 EBAG9 KCNV1<br />
TRHR GOLSYN<br />
NUDCD1<br />
3.4 Mb<br />
Fig. 1. GWAS and f<strong>in</strong>e-mapp<strong>in</strong>g identify RSPO2 as<strong>the</strong>associatedgeneformoustacheandeyebrow<br />
growth pattern (furn<strong>is</strong>h<strong>in</strong>gs). (A) Three types of coat segregate <strong>in</strong> dachshunds: (from left to right) smoothcoated,<br />
long-haired, and wire-haired with furn<strong>is</strong>h<strong>in</strong>gs. (B) Results of <strong>the</strong> GWAS <strong>in</strong> <strong>the</strong> dachshund us<strong>in</strong>g<br />
wire-haired dogs as cases and smooth-coated and long-haired dogs as controls. The best P value (3.35 ×<br />
10 −27 ), highlighted by <strong>the</strong> arrow, <strong>is</strong> located on CFA13 at position 11,095,120. (C) Results of <strong>the</strong> GWAS for<br />
furn<strong>is</strong>h<strong>in</strong>gs <strong>in</strong> <strong>the</strong> CanMap data set. The arrow highlights <strong>the</strong> best association (P =10 −241 )atCFA13<br />
position 11,659,792. Both P values were obta<strong>in</strong>ed with s<strong>in</strong>gle-marker c 2 analyses. (D) Homozygous regions<br />
identified <strong>in</strong> cases from GWAS and f<strong>in</strong>e-mapp<strong>in</strong>g. The red rectangle represents <strong>the</strong> associated haplotype <strong>in</strong><br />
<strong>the</strong> dachshund; <strong>the</strong> blue rectangle spans <strong>the</strong> homozygous region from 19 breeds fixed for furn<strong>is</strong>h<strong>in</strong>gs<br />
based on <strong>the</strong> multibreed data set; <strong>the</strong> green rectangle <strong>in</strong>dicates <strong>the</strong> region of homozygosity <strong>in</strong> 18 breeds<br />
fixed for <strong>the</strong> furn<strong>is</strong>h<strong>in</strong>gs after f<strong>in</strong>e-mapp<strong>in</strong>g. A 167-bp deletion, <strong>in</strong>dicated by <strong>the</strong> small red rectangle, <strong>is</strong><br />
located with<strong>in</strong> all three haplotypes <strong>in</strong> <strong>the</strong> 3′UTR of <strong>the</strong> RSPO2 gene. The positions of genes <strong>in</strong> <strong>the</strong> region<br />
are represented by open boxes at <strong>the</strong> bottom of <strong>the</strong> figure, labeled to <strong>the</strong> left, with arrows <strong>in</strong>dicat<strong>in</strong>g<br />
read<strong>in</strong>g-frame direction (www.genome.ucsc.edu).<br />
REPORTS<br />
<strong>the</strong> orig<strong>in</strong>al 3.4-Mb haplotype observed <strong>in</strong> <strong>the</strong><br />
dachshund-only GWAS, and a 2.8-Mb haplotype<br />
identified <strong>in</strong> crossover analys<strong>is</strong> with<strong>in</strong> <strong>the</strong><br />
dachshund pedigree (Fig. 1D).<br />
F<strong>in</strong>e-mapp<strong>in</strong>g allowed us to reduce <strong>the</strong><br />
homozygous region to 238 kb spann<strong>in</strong>g only<br />
<strong>the</strong> R-spond<strong>in</strong>–2 (RSPO2) gene, exclud<strong>in</strong>g <strong>the</strong><br />
5´ untranslated region (5′UTR) and <strong>the</strong> first<br />
exon (Fig. 1D, fig. S1, and table S3). RSPO2 <strong>is</strong><br />
an excellent candidate for a hair-growth phenotype<br />
as it synergizes with Wnt to activate bcaten<strong>in</strong><br />
(12), and Wnt signal<strong>in</strong>g <strong>is</strong> required for<br />
<strong>the</strong> establ<strong>is</strong>hment of <strong>the</strong> hair follicles (13, 14).<br />
Moreover, <strong>the</strong> Wnt/b-caten<strong>in</strong> pathway <strong>is</strong> <strong>in</strong>volved<br />
<strong>in</strong> <strong>the</strong> development of hair-follicle tumors, or pilomatricomas<br />
(15), which occur most frequently <strong>in</strong><br />
breeds that have furn<strong>is</strong>h<strong>in</strong>gs (16). Recent studies<br />
have shown that a mutation <strong>in</strong> <strong>the</strong> EDAR gene,<br />
also <strong>in</strong>volved <strong>in</strong> <strong>the</strong> Wnt pathway, <strong>is</strong> responsible<br />
for a coarse East-Asian hair type found <strong>in</strong> humans<br />
(17), with some similarity to can<strong>in</strong>e wirehair.<br />
All exons and conserved regions of RSPO2<br />
were sequenced <strong>in</strong> dogs from seven breeds (table<br />
S4). Only an <strong>in</strong>sertion of 167 base pairs (bp)<br />
with<strong>in</strong> <strong>the</strong> 3′UTR at position 11,634,766 was<br />
perfectly associated with <strong>the</strong> furn<strong>is</strong>h<strong>in</strong>gs trait <strong>in</strong><br />
dogs from both <strong>the</strong> case/control study and <strong>the</strong> extended<br />
pedigree (table S5). The result was fur<strong>the</strong>r<br />
confirmed <strong>in</strong> a set of 704 dogs of vary<strong>in</strong>g phenotypes.<br />
In total, 297 of 298 dogs with furn<strong>is</strong>h<strong>in</strong>gs<br />
were ei<strong>the</strong>r homozygous (268) or heterozygous<br />
(29) for <strong>the</strong> <strong>in</strong>sertion, and all 406 dogs lack<strong>in</strong>g<br />
<strong>the</strong> trait were homozygous for <strong>the</strong> ancestral state,<br />
as <strong>is</strong> cons<strong>is</strong>tent with a dom<strong>in</strong>ant mode of <strong>in</strong>heritance<br />
(table S1).<br />
Th<strong>is</strong> mutation does not affect <strong>the</strong> prote<strong>in</strong>cod<strong>in</strong>g<br />
region of <strong>the</strong> RSPO2 gene. However, because<br />
<strong>the</strong> 3′UTR frequently encodes elements<br />
that <strong>in</strong>fluence mRNA stability [reviewed <strong>in</strong> (18)],<br />
we exam<strong>in</strong>ed whe<strong>the</strong>r <strong>the</strong> <strong>in</strong>sertion was associated<br />
with a change <strong>in</strong> <strong>the</strong> expression level of <strong>the</strong><br />
RSPO2 gene. We found a threefold <strong>in</strong>crease <strong>in</strong><br />
RSPO2 transcripts <strong>in</strong> muzzle sk<strong>in</strong> biopsies of dogs<br />
with furn<strong>is</strong>h<strong>in</strong>gs, cons<strong>is</strong>tent with a transcript effect<br />
(fig. S2).<br />
We applied <strong>the</strong> same mapp<strong>in</strong>g strategy to<br />
hair length. Previously, mutations <strong>in</strong> <strong>the</strong> FGF5<br />
gene were identified <strong>in</strong> Welsh corg<strong>is</strong> segregat<strong>in</strong>g<br />
an atypical “fluffy” or long-haired phenotype (19)<br />
and associated with excess hair growth <strong>in</strong> mice<br />
and cats (20–22). Our study replicates <strong>the</strong>se f<strong>in</strong>d<strong>in</strong>gs<br />
<strong>in</strong> an extended breed set. Indeed, association<br />
analyses <strong>in</strong> both <strong>the</strong> dachshund and CanMap data<br />
sets highlight <strong>the</strong> region on CFA32 conta<strong>in</strong><strong>in</strong>g<br />
FGF5 with P values of 3 × 10 −27 and 9 × 10 −44 ,<br />
respectively. After f<strong>in</strong>e-mapp<strong>in</strong>g, a 67-kb homozygous<br />
region highlighted <strong>the</strong> FGF5 gene (Fig.<br />
2A, fig. S3, and table S6). The strongest association<br />
was observed at position 7,473,337 (P =<br />
1×10 −157 ), <strong>in</strong> which a highly conserved Cys<br />
<strong>is</strong> changed to Phe (Cys 95 →Phe) <strong>in</strong> exon 1 of<br />
FGF5, cons<strong>is</strong>tent with <strong>the</strong> previous study (19).<br />
Sequenc<strong>in</strong>g with<strong>in</strong> <strong>the</strong> homozygous haplotype<br />
revealed no SNPs with stronger association<br />
(table S7).<br />
www.sciencemag.org SCIENCE VOL 326 2 OCTOBER 2009 151<br />
on June 29, 2010<br />
www.sciencemag.org<br />
Downloaded from





