

Natural Medicine Law - Dietary Supplement Regulation, Herbal ...
Natural Medicine Law - Dietary Supplement Regulation, Herbal ...
Natural Medicine Law - Dietary Supplement Regulation, Herbal ...

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
Vol. 10, No. 6 May 2007<br />
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM Newsletter<br />
GMP REGULATIONS<br />
STILL AT OMB<br />
OMB still has the <strong>Dietary</strong><br />
<strong>Supplement</strong> Good Manufacturing<br />
Practice regulations and has not<br />
sent them to FDA as reported in<br />
other news articles you may have<br />
read. FDA spokesperson Linda<br />
Kahl in College Park, Maryland,<br />
where the new headquarters of the<br />
Center for Food Safety and Applied<br />
Nutrition is located, told NML on<br />
April 30 that the White House<br />
Office of Management and Budget<br />
has not signed off on the<br />
regulations.<br />
The regulations were first<br />
requested in October 1994 when<br />
DSHEA was passed, but the FDA<br />
did not submit a draft until<br />
February 1997. After getting<br />
comments and revising the<br />
proposal, the FDA published a new<br />
version in March of 2003 in the<br />
Federal Register. Now, four years<br />
later, there is nothing much<br />
happening.<br />
The Federal government has set<br />
up a new website to keep track of<br />
regulatory processes. You can go<br />
to: www.reginfo.gov then to the<br />
Department of Health and Human<br />
Services to find what is pending,<br />
including GMPs. There we are told<br />
there is “no” legal deadline or<br />
timetable for adopting GMPs for<br />
dietary supplements. HHS<br />
classifies the regulations as<br />
“major” and says they will have a<br />
“significant economic impact.”<br />
HHS (apparently on FDA’s best<br />
EPHEDRA APPEAL<br />
Statutory interpretation is what the<br />
case is all about. FDA and the 10 th<br />
Circuit read the law one way;<br />
Nutraceutical International<br />
Corporation and its attorneys read<br />
the FDC Act another way. A<br />
Petition for Certiorari has been<br />
filed with the Supreme Court of the<br />
United States by Nutraceutical.<br />
The <strong>Natural</strong> Products Association<br />
has filed an Amicus Curiae petition<br />
in support of accepting certiorari.<br />
The Justices will read the petitions<br />
and determine if they want to hear<br />
the case or not. [See next column.]<br />
If the Petition is denied, the 10 th<br />
Circuit opinion and orders will be<br />
the law of the land, and the 10 mg<br />
of ephedra alkaloids in capsules are<br />
prohibited from sale in the United<br />
States, while the same and higher<br />
doses are still permitted in food and<br />
traditional Asian tea formulations.<br />
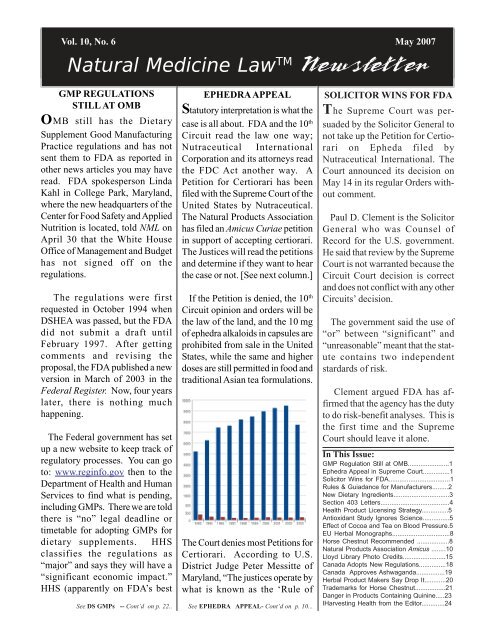
The Court denies most Petitions for<br />
Certiorari. According to U.S.<br />
District Judge Peter Messitte of<br />
Maryland, “The justices operate by<br />
what is known as the ‘Rule of<br />
See DS GMPs -- Cont’d on p. 22.. See EPHEDRA APPEAL- Cont’d on p. 10...<br />
SOLICITOR WINS FOR FDA<br />
The Supreme Court was persuaded<br />
by the Solicitor General to<br />
not take up the Petition for Certiorari<br />
on Epheda filed by<br />
Nutraceutical International. The<br />
Court announced its decision on<br />
May 14 in its regular Orders without<br />
comment.<br />
Paul D. Clement is the Solicitor<br />
General who was Counsel of<br />
Record for the U.S. government.<br />
He said that review by the Supreme<br />
Court is not warranted because the<br />
Circuit Court decision is correct<br />
and does not conflict with any other<br />
Circuits’ decision.<br />
The government said the use of<br />
“or” between “significant” and<br />
“unreasonable” meant that the statute<br />
contains two independent<br />
stardards of risk.<br />
Clement argued FDA has affirmed<br />
that the agency has the duty<br />
to do risk-benefit analyses. This is<br />
the first time and the Supreme<br />
Court should leave it alone.<br />
In This Issue:<br />
GMP <strong>Regulation</strong> Still at OMB.......................1<br />
Ephedra Appeal in Supreme Court...............1<br />
Solicitor Wins for FDA..................................1<br />
Rules & Guiadance for Manufacturers.........2<br />
New <strong>Dietary</strong> Ingredients...............................3<br />
Section 403 Letters......................................4<br />
Health Product Licensing Strategy...............5<br />
Antioxidant Study Ignores Science...............5<br />
Effect of Cocoa and Tea on Blood Pressure.5<br />
EU <strong>Herbal</strong> Monographs................................8<br />
Horse Chestnut Recommended ..................8<br />
<strong>Natural</strong> Products Association Amicus ........10<br />
Lloyd Library Photo Credits........................15<br />
Canada Adopts New <strong>Regulation</strong>s...............18<br />
Canada Approves Ashwaganda................19<br />
<strong>Herbal</strong> Product Makers Say Drop It............20<br />
Trademarks for Horse Chestnut.................21<br />
Danger in Products Containing Quinine.....23<br />
IHarvesting Health from the Editor.............24
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside<br />
Page 2 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
RULES AND GUIDANCE<br />
FOR<br />
MANUFACTURERS AND<br />
DISTRIBUTORS<br />
For the first time, the United<br />
Kingdom’s Crown Printer has<br />
allowed the Royal Pharmaceutical<br />
Society of Great Britain to take over<br />
the publication of documents that<br />
were always before published only by<br />
the government printer. The Crown<br />
still owns the copyright. Rules and<br />
Guidance for Pharmaceutical<br />
Manufacturers and Distributors is a<br />
paper or computer loadable program<br />
that interacts with the latest<br />
information on the web from the U.K.<br />
government.<br />
NML is telling readers about this<br />
because some of you are interested in<br />
the regulation and manufacture of<br />
herbal medicines in the U.K. and the<br />
E.C. And those readers of NML who<br />
sell or prescribe herbal medicines<br />
need to know what the governments<br />
in Europe are asking be done. Since<br />
1971 this publication has been known<br />
as the “Orange Guide.” Now it has<br />
become digital for the first time.<br />
When you download the disk to<br />
your computer and open it up, you get<br />
documents that were compiled by the<br />
Inspection and Standards Division,<br />
<strong>Medicine</strong>s and Healthcare products<br />
Regulatory Agency (MHRA),<br />
London, UK. The contents are listed<br />
as:<br />
I MEDICINES AND HEALTHCARE<br />
PRODUCTS REGULATORY<br />
AGENCY (MHRA)<br />
1 MHRA: Licensing, Inspection and<br />
Enforcement for Human <strong>Medicine</strong>s<br />
II GUIDANCE ON GOOD<br />
MANUFACTURING PRACTICE<br />
(GMP)<br />
The MHRA Index to the EU Guide to<br />
GMP<br />
2 EU Guidance on Good<br />
Manufacturing Practice<br />
3 UK Guidance on Manufacture<br />
III LEGISLATION ON<br />
MANUFACTURE<br />
4 EU Legislation on Manufacture<br />
5 UK Legislation on Manufacture<br />
IV GUIDANCE ON WHOLESALE<br />
DISTRIBUTION PRACTICE<br />
6 EU Guidance on Wholesale<br />
Distribution Practice<br />
7 UK Guidance on Wholesale<br />
Distribution Practice<br />
V LEGISLATION ON WHOLESALE<br />
DISTRIBUTION<br />
8 EU Legislation on Wholesale<br />
Distribution<br />
9 UK Legislation on Wholesale<br />
Distribution<br />
VI GLOSSARY OF LEGISLATION<br />
Glossary of Legislation<br />
In these six sections and nine<br />
chapters, you get some common sense<br />
information that needs to be said to<br />
clear the air. And the program<br />
provides a search engine to find such<br />
items as “<strong>Herbal</strong> <strong>Medicine</strong>” that is<br />
covered in Annex 7A where it states,<br />
for example, under Premises:<br />
“Storage areas<br />
“1 Crude (i.e. unprocessed) plants<br />
should be stored in separate areas. The<br />
storage area should be well ventilated<br />
and be equipped in such a way as to<br />
give protection against the entry of<br />
insects or other animals, especially<br />
rodents. Effective measures should be<br />
taken to prevent the spread of any<br />
such animals and micro-organisms<br />
brought in with the crude plant and<br />
to prevent cross-contamination.<br />
Containers should be located in such<br />
a way as to allow free air circulation.<br />
“2 Special attention should be paid<br />
to the cleanliness and good<br />
See U.K. RULES -- Continued on page 17...<br />
Search old issues on the Web<br />
WWW.NATMEDLAW.COM<br />
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
Newsletter<br />
Muscatatuck Publishers, Inc.<br />
P.O. Box 741261<br />
Boynton Beach, FL 33474-1261<br />
William J. Skinner<br />
Editor and Publisher,<br />
Registered Pharmacist and Attorney<br />
Copyright 2007 by Muscatatuck Publishers, Inc.<br />
Exclusive of U.S. Government document excerpts,<br />
photocopying, faxing, and electronic transfer is prohibited<br />
without permission. Copies of all documents<br />
mentioned in this publication, except copyrighted<br />
materials, are available on request for a fee. Telephone<br />
(866) 664-2900 or (561) 641-2900 or E-mail<br />
at: for permission.<br />
Published six times a year. Subscription price is<br />
US$249.00 per year in U.S., Canada, and Mexico.<br />
Subscriptions to other countries are an additional<br />
US$24.00 per year for postage and handling. Additional<br />
copy in same envelope for US$130.00.<br />
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> (ISSN 1095-6336) reports<br />
on legal issues. Readers should discuss legal issues<br />
with legal counsel before taking action based on<br />
information in this publication. Subscribers registered<br />
with the Copyright Clearance Center may reproduce<br />
parts or all of <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> for<br />
educational classes, provided that a base fee is paid<br />
directly to Copyright Clearance Center, 222 Rosewood<br />
Drive, Danvers, MA 09123, telephone (508)<br />
750-8400. Violation of copyright will result in legal<br />
action, including civil and criminal penalties<br />
and suspension of service.<br />
Classified Ads<br />
Deadline for each issue is the 1st day of month published,<br />
January, March, May, July, September, and<br />
November.<br />
Classified Rates<br />
Minimum of $30.00 for 20 words. Additional words<br />
are $1.00 each. Name and address are included in<br />
word count. Telephone numbers are one word. State<br />
and zip code count as one word. Special headings<br />
larger than 12 point type are $2.00 per word. Capitalized<br />
or italicized wording no extra charge. Send<br />
advertisement copy to Muscatatuck Publishers, Inc,<br />
address above. Payment must accompany ad copy.<br />
Display Ads<br />
A limited number of display ads may be featured in<br />
each issue. Subscribers are attorneys, health care<br />
practitioners, company officers, research scientists,<br />
university faculty members, and herbalists. Subscribers<br />
are located throughout the world. New<br />
groups are provided one or more free sample issues<br />
per year in promotions at various meetings and<br />
through direct mail. Prices are available on the<br />
website under Advertising Information.<br />
Editorial Correspondence<br />
Florida address above<br />
Toll-Free Telephone: (866) 664-2900<br />
E-mail: natmedlaw@AOL.com
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 3<br />
NEW DIETARY<br />
INGREDIENTS<br />
The summaries that NML prints<br />
here are condensations of the<br />
notices filed for new dietary<br />
ingredients (NDIs) by<br />
manufacturers and distributors.<br />
Each manufacturer and distributor<br />
that wants to legally market a NDI<br />
must make a separate submission.<br />
The regulations do not allow for<br />
piggybacking on other company’s<br />
notices. Since the DSHEA was<br />
signed in 1994, and Docket No.<br />
95S-0316 was opened in 1995,<br />
FDA finally cleared up the process<br />
for filing Section 413(a) notices<br />
only ten years later. There are still<br />
less than 400 FDA reports reviewed<br />
and filed in this public dockets at<br />
FDA. The agency granted two<br />
NDIs, one for ADM’s BeneFlax<br />
Flax Lignan Extract and the other<br />
for the NDI of Iwade Research for<br />
Freeze-Dried Extract of Agaricus<br />
blazei Murrill.<br />
SEPPIC, INC. of Fairfield,<br />
New Jersey filed a notice of a New<br />
<strong>Dietary</strong> Ingredient (NDI) by letter<br />
on June 8, 2006, amending its<br />
earlier notice letter of April 28,<br />
2006, for EXTRAMEL ® ,<br />
containing Cantalope melon<br />
(cucumis Melo L – Carl Linneaus)<br />
extract rich in antioxidants.<br />
EXTRAME ® L is recommended for<br />
use as 10 mg daily dose, equivalent<br />
to 142 I.U. of antioxidant enzymes<br />
and 1.8 ì g of other antioxidants as<br />
vitamins A, E, C, carotenoids. This<br />
dose contains less than the ordinary<br />
daily serving of cantaloupe and<br />
may be considered entirely safe,<br />
says the notice. In EXTRAMEL ®<br />
the extract is coated with 80% of<br />
hydrogenated palm oil to protect<br />
the antioxidants. The extract is<br />
concentrated from crushed<br />
cantalope melon, centrifuged and<br />
filtered, then concentrated. The<br />
result is freeze dried and coated<br />
with palm oil as a protective<br />
excipient. Rinds, seeds and pulp<br />
are not included. No chemicals or<br />
catalysts are added. One oral<br />
toxicity test in rats was included.<br />
FDA responded by letter, dated<br />
July 20, 2006, from Linda S.<br />
Pellicore, Ph.D., stating that the<br />
agency had concerns about the<br />
evidence provided because the<br />
extraction process was only<br />
described in very general terms and<br />
did not provide manufacturing<br />
details. Also, the notice did not<br />
provide a history of use, whole<br />
cantalope, including the rind,<br />
establishing that when used as<br />
suggested will reasonably be<br />
expected to be safe. For these<br />
reasons, the information provided<br />
does not provide an adequate basis<br />
to conclude that EXTRAMEL ®<br />
will reasonably be expected to be<br />
safe. Therefore, the product may<br />
be adulterated and introduction of<br />
the product in interstate commerce<br />
is prohibited. Docket No. 1995S-<br />
0316, RPT 353, received at the<br />
Dockets Office on August 18,<br />
2006, filed August 18, 2006, and<br />
posted to the FDA Website on<br />
November 20, 2006 by kk.<br />
<strong>Natural</strong> ASA of Lysaker,<br />
Norway, filed an NDI notice, dated<br />
May 15, 2006, for its product<br />
Omega-3 Phospholipids, containing<br />
in each gram 105 to 135 mg<br />
of EPA and 50 to 75 mg of DHA,<br />
and 155 to 210 mg combined<br />
EPA+DHA, with a DPA:DHA<br />
ration of approximately 2:1. The<br />
recommended use is the<br />
consumption of up to 4 g per day.<br />
An attachment concerning safety<br />
was provided that included<br />
sections on regulatory status,<br />
background exposure from diet,<br />
metabolism, animal studies,<br />
clinical studies, and a summary.<br />
This attachment was 83 pages of<br />
information, some parts of which<br />
were redacted. FDA took until<br />
August 4, 2006 to respond by letter<br />
to Egil Nilsen from Linda S.<br />
Pellicore, Ph.D. stating that the<br />
agency was concerned about the<br />
evidence upon which the company<br />
relies to conclude that the product<br />
will reasonably be expected to be<br />
safe. FDA did not know how many<br />
drops, milliliters, teaspoons or<br />
similar units of liquid measure will<br />
be used to measure the 4 g daily<br />
serving. And FDA said the 83<br />
pages did not include the safety of<br />
the components of the product or<br />
the entire mixture. Therefore, FDA<br />
said it is unclear how the<br />
interesterfied components are<br />
qualitatively or quantitatively<br />
similar to the substances described<br />
in the safety information in the<br />
notification or how that<br />
information is relevant to the<br />
product. For these reasons, FDA<br />
said the product may be adulterated<br />
under the Act and introduction of<br />
such a product into interstate<br />
commerce is prohibited. Docket<br />
No. 1995S-0316, RPT 354,<br />
received at the Dockets Office on<br />
October 6, 2006, filed October 6,<br />
2006, and posted to the FDA<br />
Website on November 20, 2006 by<br />
kk.<br />
Arthur Daniels Midland<br />
Company of Decatur, Illinois sent<br />
a NDI notice dated June 6, 2006 to<br />
See NEW INGREDIENTS -- Cont’d on p. 7...<br />
www.NatMed<strong>Law</strong>.com<br />
for<br />
<strong>Natural</strong> <strong>Medicine</strong> Ties TM<br />
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside<br />
Page 4 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
SECTION 403 LETTERS<br />
More “Courtesy Letters” or<br />
“Letters of Rejection” are being<br />
filed by FDA staff in response to<br />
notices of structure/function claims<br />
or statements of health claims for<br />
dietary supplements. Fewer than<br />
one in twenty notices to FDA gets<br />
such a response, but FDA has<br />
reviewed nearly 20,000 notices so<br />
far. Here are the latest Section 403<br />
letters.<br />
Enzymatic Therapy, Inc. of<br />
Green Bay, Wisconsin, filed a<br />
notice, dated November 8 and 10,<br />
2006 to the FDA for the products<br />
Liquid Calcium Magnesium with<br />
Botanicals and ITI Mega Multi<br />
Vitamin Drink Mix. The Liquid<br />
with Botanicals product contains<br />
Vitamin D, calcium carbonate,<br />
magnesium oxide and boron oxide,<br />
and the amounts are not disclosed<br />
in the filing. The are six claims:<br />
“Provides synergistic boneenhancing<br />
nutrients,” “Bone<br />
health is built on calcium … and<br />
more!,” “Liquid Calcium<br />
Magnesium with Botanicals<br />
maintains optimum bone health<br />
and may reduce the risk of<br />
osteoporosis,” “ Vitamin D and<br />
boron promote absorption of<br />
calcium,” “Giving you bones what<br />
they need has never been easier …<br />
or more delicious!,” and<br />
“Features a highly concentrated<br />
and bioavailable form of calcium<br />
plus magnesium.” For the Drink<br />
Mix product there are a long list of<br />
vitamins, minerals, amino acids,<br />
and botanicals. The single claim<br />
being made is: “Combined with<br />
additional ingredients, including<br />
natural fruit extract antioxidants<br />
and amino acids, they can provide<br />
nutritional assistance for optimal<br />
energy and healthy stress<br />
management.” FDA’s Vasilios H.<br />
Frankos, Ph.D. responded to<br />
Michael P. Devereaux by letter of<br />
November 28, 2006 stating that the<br />
claim concerning risk of<br />
osteoporosis is one that is subject<br />
to 21 CFR 101.72 and failure to<br />
make a claim in accordance with<br />
that regulation subjects the product<br />
to regulation as a drug. Also, the<br />
FDA letter said that a Drink Mix is<br />
not a “dietary supplement” under<br />
the Act, but is a conventional food<br />
that must meet the food<br />
regulations. Ingredients in food<br />
must be GRAS or approved food<br />
additives or the product is<br />
adulterated and cannot be marketed<br />
legally in the U.S. Docket No.<br />
97S-0163, Ltr 911, received at<br />
Dockets on December 18, 2006,<br />
entered the Docket on December<br />
22, 2006 and posted on the FDA<br />
Website on January 10, 2007.<br />
Flora, Inc. of Lynden,<br />
Washington, wrote FDA two letters<br />
on November 3, 2006 to give<br />
notice that it was filing certain<br />
claims for the products Vegetarian<br />
DHA Flax Oil <strong>Dietary</strong> <strong>Supplement</strong><br />
and Udo’s DHA Oil Blend<br />
<strong>Dietary</strong> <strong>Supplement</strong>. Ingredients<br />
of both products were omitted from<br />
the letters. The claims for the Flax<br />
Oil product were: “Supports<br />
cognitive function, heart health,<br />
and eye health,” “DHA is an<br />
omega-3 fatty acid that is essential<br />
for the proper functioning of our<br />
brains as adults, and for the<br />
development of our nervous system<br />
and visual abilities during the first<br />
six months of life,” and<br />
“Supportive but not conclusive<br />
research shows that consumption<br />
of EPA and DHA omega-3 fatty<br />
acids may help reduce the risk of<br />
coronary heart disease.” For the<br />
DHA Blend product, the claims<br />
Digitalis<br />
Photo Courtesy of Lloyd Library and Museum<br />
were the same. FDA responded to<br />
Nancy Blake, on November 28,<br />
2006, by letter from Vasilios H.<br />
Frankos, Ph.D. stating that the<br />
claim that the products will reduce<br />
the risk of heart disease is a disease<br />
claim that they will treat coronary<br />
heart disease and if these claims<br />
are used, these products will be<br />
regulated as drug products under<br />
the Act. FDA added that failure to<br />
make claims that are not in<br />
accordance with certain<br />
enforcement letters with respect to<br />
claims about reduced risk of<br />
coronary heart disease at<br />
www.cfdan.fda.gov/~dms/labqhc.html<br />
would misbrand the<br />
dietary supplement and make the<br />
product subject to regulation as a<br />
drug under the Act. Docket No.<br />
97S-0163, Ltr. 912, received at<br />
Dockets December 18, 2006,<br />
entered into the Docket on<br />
December 22, 2006, and posted to<br />
the FDA Website on January 10,<br />
2007.<br />
See SECTION 403 -- Continued on page 14...
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 5<br />
NATURAL HEALTH<br />
PRODUCT<br />
LICENSING STRATEGY<br />
Health Canada announced April<br />
10, 2007 that it wants clarify the<br />
product licensing application<br />
process (i.e. the process for<br />
obtaining a NPN) for those natural<br />
health products (NHPs) currently<br />
holding Drug Identification<br />
Numbers (DIN) issued prior to<br />
January 1, 2004. Specifically, this<br />
document clarifies that the<br />
application process for NHPs with<br />
DINs consists of a regular Product<br />
License Application without the<br />
safety and efficacy information<br />
required in section 5(g), as<br />
authorized under Sections 108 and<br />
109 of the <strong>Natural</strong> Health Products<br />
<strong>Regulation</strong>s.<br />
As outlined in the transition<br />
provisions of the <strong>Natural</strong> Health<br />
Products <strong>Regulation</strong>s, all NHPs<br />
with DINs must obtain a valid<br />
product license and <strong>Natural</strong><br />
Product Number (NPN) no later<br />
than December 31, 2009.<br />
Applicants are encouraged to apply<br />
as early as possible to transition<br />
their DINs to NPNs. This will<br />
Big Ben and Parliament in London<br />
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> Around the World<br />
See STRATEGY -- Continued on page 6...<br />
ANTIOXIDANT STUDY<br />
IGNORES SCIENTIFIC<br />
EVIDENCE<br />
A meta-analysis undermining the<br />
benefits of antioxidant supplements<br />
in a new study published in the<br />
February 28, 2007 issue of the<br />
Journal of the American Medical<br />
Association ignores the large body<br />
of scientific evidence supporting the<br />
benefits of antioxidants, says the<br />
Canadian Health Food Association,<br />
a national trade association<br />
representing Canada’s natural<br />
products industry.<br />
“A growing number of evidencebased<br />
studies continue to show the<br />
health benefits of antioxidants and<br />
cannot be discounted. They need to<br />
be considered when drawing<br />
conclusions from this metaanalysis,”<br />
says Anne Wilkie, Vice-<br />
President and Head of Regulatory<br />
Affairs, Canadian Health Food<br />
Association (CHFA.)<br />
“The study only serves to confuse<br />
healthy consumers who may take<br />
antioxidants,” she says. The<br />
researchers themselves<br />
acknowledge their analysis of<br />
previous studies has several<br />
limitations, including combining<br />
studies where participants were<br />
vastly different from each other<br />
(healthy versus diseased<br />
populations); where dosages used<br />
varied significantly; where the<br />
length of time of taking the<br />
supplements and the follow-up<br />
differed among the trials, and the use<br />
of varying definitions of “all-cause”<br />
mortality in the trials. Meta analyses<br />
are valuable tools when the included<br />
studies are similar in design and<br />
study populations.<br />
See CHFA -- Continued on page 18...<br />
EFFECT OF<br />
COCOA AND TEA<br />
ON BLOOD PRESSURE<br />
This abstract is from the Arch<br />
Intern Med. 2007;167:626-634,<br />
April 9, 2007 by authors Dirk<br />
Taubert, MD, PhD; Renate Roesen,<br />
PhD; and Edgar Schömig, MD, all<br />
of the Department of<br />
Pharmacology, University Hospital<br />
of Cologne, Cologne, Germany.<br />
This was a meta-analysis of<br />
randomized controlled trials to<br />
determine changes in systolic and<br />
diastolic blood pressure due to the<br />
intake of cocoa products or black<br />
and green tea. Studies from<br />
MEDLINE, EMBASE, SCOPUS,<br />
Science Citation Index, and the<br />
Cochrane Controlled Trials<br />
Register were searched from 1966<br />
until October 2006. The authors<br />
found studies in parallel group or<br />
crossover design involving 10 or<br />
more adults in whom blood<br />
pressure was assessed before and<br />
after receiving cocoa products or<br />
black or green tea for at least 7<br />
days.<br />
There were five cocoa studies<br />
and five teas studies. Five<br />
randomized controlled studies of<br />
cocoa administration involving a<br />
total of 173 subjects with a median<br />
duration of 2 weeks were included.<br />
After the cocoa diets, the pooled<br />
mean systolic and diastolic blood<br />
pressure were –4.7 mm Hg (95%<br />
confidence interval [CI], –7.6 to –<br />
1.8 mm Hg; P = .002) and –2.8 mm<br />
Hg (95% CI, –4.8 to –0.8 mm Hg;<br />
P = .006) lower, respectively,<br />
compared with the cocoa-free<br />
controls. Five studies of tea<br />
consumption involving a total of<br />
See COCOA IN HBP -- Cont’d on page 8...<br />
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside<br />
Page 6 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
STRATEGY -- Continued from page 5...<br />
provide the <strong>Natural</strong> Health<br />
Products Directorate (NHPD) with<br />
adequate time to review the<br />
application and will avoid any<br />
interruption in the sale of their<br />
products.<br />
Section 109 of the <strong>Natural</strong><br />
Health Products <strong>Regulation</strong>s<br />
creates an exemption from the<br />
safety and efficacy requirements in<br />
Section 5(g) for products<br />
previously assessed and licensed<br />
by the Therapeutic Products<br />
Directorate (TPD) under the Food<br />
and Drug <strong>Regulation</strong>s (i.e., natural<br />
health products previously granted<br />
market authorization pursuant to a<br />
DIN).<br />
NHPs with DINs must receive a<br />
NPN by December 31, 2009.<br />
Companies may seek an NPN for<br />
their NHPs with DINs by<br />
submitting a Transitional DIN<br />
Product Application to the NHPD<br />
for assessment. The NHPD will<br />
review these transitional<br />
applications to ensure that the<br />
products comply with the <strong>Natural</strong><br />
Health Products <strong>Regulation</strong>s.<br />
During assessment, the NHPD<br />
will focus on the elements required<br />
by the <strong>Natural</strong> Health Products<br />
<strong>Regulation</strong>s once the exemption in<br />
Section 109 is applied to these<br />
applications. These elements<br />
include source material, nonmedicinal<br />
ingredients (NMIs) and<br />
finished product specifications.<br />
Information on the label, where the<br />
<strong>Natural</strong> Health Products<br />
<strong>Regulation</strong>s requirements differ<br />
from the requirements under the<br />
Food and Drug <strong>Regulation</strong>s, will<br />
also be addressed to ensure<br />
compliance with the labeling<br />
requirements for NHPs.<br />
Applicants will only be able to<br />
file a Transitional DIN Product<br />
Application if the product and<br />
associated claims are identical to<br />
what was approved by the TPD.<br />
Any changes to the product and/or<br />
associated claims will result in the<br />
removal of the submission from<br />
this process / refusal of the<br />
submission. Note the only<br />
exceptions: addition of risk<br />
information to the product (e.g. risk<br />
statements from NHPD<br />
monographs) and changes to nonmedicinal<br />
ingredients.<br />
Prior to the issuance of a Product<br />
License and NPN for NHPs with<br />
DINs, the NHPD requires all<br />
products to meet the requirements<br />
of the <strong>Natural</strong> Health Products<br />
<strong>Regulation</strong>s. Applicants are<br />
encouraged to include new risk<br />
information from published NHPD<br />
monographs when filing a<br />
Transitional DIN Product<br />
Application. This additional<br />
information allows the consumer to<br />
make a more informed decision<br />
about a product.<br />
Transitional DIN Product<br />
Applications must include all the<br />
information listed further below<br />
(Transitional DIN Application<br />
Requirements). The NHPD will<br />
conduct a review of the application<br />
to ensure that all application<br />
requirements have been met. A<br />
Product Licence and NPN will be<br />
issued in which the recommended<br />
conditions of use (i.e. claims, dose,<br />
duration of use, dosage form, and<br />
risk information) are listed.<br />
Should an applicant wish to<br />
submit amendments not previously<br />
approved by the TPD with their<br />
application, the application will be<br />
assessed in the applicable Non-<br />
Transitional DIN queue.<br />
Transitional DIN Application<br />
Requirements<br />
As of May 1, 2007, all<br />
Transitional DIN Product<br />
Applications received by the<br />
NHPD must include the<br />
information listed below or the<br />
application may be refused. For<br />
transitional applications currently<br />
in queue at the NHPD that do not<br />
meet the requirements listed below,<br />
the applicants will be contacted by<br />
the NHPD and given the<br />
opportunity to amend their<br />
applications accordingly.<br />
1. Cover Letter including<br />
the following attestations:<br />
o<br />
o<br />
o<br />
o<br />
the DIN is valid at<br />
the time of filing;<br />
the product and<br />
label are in<br />
compliance with<br />
the terms of<br />
marketing<br />
authorization<br />
issued by the TPD<br />
under the Food &<br />
Drug <strong>Regulation</strong>s;<br />
the product and<br />
associated claims<br />
are identical to<br />
what was<br />
previously<br />
approved by the<br />
TPD; and<br />
the NMIs are listed<br />
on the NHPD’s List<br />
of Acceptable<br />
NMIs, otherwise<br />
the cover letter<br />
must note those not<br />
on the list;<br />
See STRATEGY -- Continued on page 11...
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 7<br />
NEW INGREDIENTS -- Continued from p. 3...<br />
FDA for the product BeneFlax<br />
Flax Lignan Extract. The notice<br />
was a new revision of a notice<br />
previously sent on December 2,<br />
2005. The letter accompanying the<br />
notice said the company had a<br />
telephone conference with Linda<br />
Pellicore, Ph.D. and Vicky Lutwak<br />
in February 2006 and it believed<br />
that this notice “clearly establish<br />
(es) that BeneFlax Flax Lignan<br />
Extract will reasonably be<br />
expected to be safe….” Attached<br />
to the letter was a package of<br />
documents bearing page numbers<br />
stamped on that indicated more<br />
than 400 pages were submitted.<br />
The initial page was marked<br />
“Confidential” and indicates it was<br />
compiled by James S. How, Ph.D.<br />
and that Luis S. Mejia, Ph.D.<br />
was to be contacted. A large<br />
number of pages were redacted;<br />
however, within the package was<br />
information stating that ADM<br />
would manufacture tablets or<br />
capsules of doses ranging from 30<br />
to 300 mg of SDG<br />
(secoisolariciresinol diglucoside)<br />
as the active ingredient which will<br />
be provided by approximately 86<br />
to 860 mg of BeneFlax Flax<br />
Lignan Extract. Also, the<br />
suggested daily dose was 86 to<br />
1720 mg of BeneFlax Flax<br />
Lignan Extract. Depending on the<br />
tablet or capsule size the suggested<br />
daily use will be “Take 1 to 4<br />
tablets or capsules a day.” SDG is<br />
the active and major lignan<br />
component. The chemical name is<br />
â-D-Glucopyranoside, 2,3-bis[(4-<br />
hydroxy-3-methoxyphenyl methyl]-1,4-butanediyl<br />
bis-,[R-)R*,<br />
R*)]. The information stated that<br />
the documents contained a literature<br />
review summary of 44 key<br />
human and animal studies in an appendix,<br />
and this is in the public<br />
record as an attachment to a letter<br />
from ADM dated June 23, 2006.<br />
FDA’s newest response to ADM<br />
was addressed to Luis J. Mejier,<br />
Ph.D. from Linda S. Pellicore,<br />
Ph.D. and dated August 18, 2006.<br />
In this letter FDA had no further<br />
questions and the agency said<br />
“FDA reviews this information to<br />
determine whether it provides an<br />
adequate basis for such a conclusion<br />
[that the evidence indicates the<br />
product when used as suggested<br />
will reasonably be expected to be<br />
safe]. Under 21 USC 350b(a)(2),<br />
there must be a history of use or<br />
other evidence of safety establishing<br />
that the new dietary ingredient,<br />
when used under the conditions<br />
recommended or suggested in the<br />
labeling of the dietary supplement,<br />
will reasonably be expected to be<br />
safe. If this requirement is not met,<br />
the dietary supplement is considered<br />
to be adulterated under 21<br />
USC 342(f)(1)(B) because there is<br />
inadequate information to provide<br />
a reasonable assurance that the new<br />
dietary ingredient does not present<br />
a significant or unreasonable risk<br />
of illness or injury. This letter is to<br />
alert you within the 75-day notification<br />
period that FDA intends to<br />
complete its evaluation within a<br />
few weeks and send you a response<br />
to your notification. Please note<br />
that a lack of a response to a notification<br />
within the 75-day<br />
timeframe does not constitute a<br />
finding by the agency that the ingredient<br />
or a product that contains<br />
the ingredient is safe or is not adulterated<br />
under 21 USC 342, See 21<br />
CFR 190.6(f).” [Because there is<br />
no other entry in this docket, and<br />
that multiple exchanges of information<br />
had taken place between<br />
ADM and FDA before these documents<br />
were placed in the public<br />
file, it is presumed that this NDI<br />
was approved. Nevertheless,<br />
FDA’s preferred legalese in handling<br />
responses to these filings<br />
leaves matters confusing and unclear.<br />
– Ed.] Docket No. 1995S-<br />
0316, RPT 355, received at the<br />
Dockets Office on October 6,<br />
2006, filed October 6, 2006, and<br />
posted to the FDA Website on November<br />
20, 2006 by kk.<br />
IWADE RESEARCH<br />
INSTITUTE OF MYCOLOGY<br />
CO., LTD, of Mie, Japan, sent<br />
FDA a NDI notice, dated June 21,<br />
2006, for its product Freeze-Dried<br />
Extract of Agaricus blazei Murrill<br />
(ABM-FD), which had been<br />
submitted in 1999 (RPT 49) and<br />
2000 (RPT 76). The cover letter<br />
points out that a very similar NDI<br />
was submitted by Japan Applied<br />
Microbiology Research Institute<br />
Ltd as RPT 276. The product is<br />
made from Himematsutake. It is<br />
to be used by dissolving the extract<br />
in water in a marked bottle,<br />
shaking, and drinking on an empty<br />
stomach prior to a meal. Each<br />
bottle contains 3.0 grams of ABM-<br />
FD. Adults (non-pregnant and<br />
non-lactating) are instructed to<br />
consume one bottle per day. The<br />
product is sold in boxes containing<br />
10 bottles. There were seven tabs<br />
of materials attached to the letter<br />
and the manufacturing information<br />
was marked “confidential.” FDA<br />
responded to Toshimitsu Sumiya<br />
by letter dated September 11, 2006<br />
from Linda S. Pellicore, Ph.D.,<br />
signed by Vasilois H. Frankos,<br />
Ph.D. stating: “FDA reviews this<br />
information to determine whether<br />
it provides an adequate basis for<br />
such a conclusion [that the<br />
evidence indicates the product<br />
when used as suggested will<br />
See NEW INGREDIENTS -- Cont’d on p. 9...<br />
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside<br />
Page 8 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
Theobroma cacao, L.<br />
Photo Courtesy of Ministry of<br />
Agriculture, Kerala State, India<br />
COCOA IN HBP -- Cont’d from page 5...<br />
343 subjects with a median<br />
duration of 4 weeks were selected.<br />
The tea intake had no significant<br />
effects on blood pressure. The<br />
estimated pooled changes were 0.4<br />
mm Hg (95% CI, –1.3 to 2.2 mm<br />
Hg; P = .63) in systolic and –0.6<br />
mm Hg (95% CI, –1.5 to 0.4 mm<br />
Hg; P = .38) in diastolic blood<br />
pressure compared with controls.<br />
The authors conclude that these<br />
randomized dietary studies indicate<br />
that consumption of foods rich in<br />
cocoa may reduce blood pressure,<br />
while tea intake appears to have no<br />
effect.<br />
EU HERBAL MONOGRAPHS<br />
The EU <strong>Herbal</strong> Monograph<br />
system is moving ahead. On<br />
March 20, 2007, the Committee on<br />
<strong>Herbal</strong> Medicinal Products<br />
published its list of status levels for<br />
herbs under consideration. The<br />
process starts with assigning a<br />
Rapporteur, then a groups<br />
discusses the information collected<br />
and publishes a draft monograph.<br />
The monograph is given an<br />
assessment and then a final opinion<br />
is adopted. The monograph then<br />
goes up the ladder to other<br />
approvals in the EU system.<br />
Final opinion adopted<br />
Aloe<br />
Frangulae cortex<br />
Lini semen Althaeae radix<br />
Plantaginis ovatae semen<br />
Plantaginis ovatae seminis<br />
tegumentum<br />
Psyllii semen<br />
Sennae folium<br />
Sennae fructus<br />
Valerianae radix<br />
Rapporteur assigned<br />
Absinthii herba<br />
Allii sativi bulbus (new Rapporteur to be<br />
appointed)<br />
Althaeae radix<br />
Avena sativa<br />
Boldi folium<br />
Calendulae flos<br />
Carvi fructus<br />
Centaurii herba<br />
Cimicifugae rhizoma<br />
Curcumae longae rhizome<br />
Cynarae folium<br />
Echinaceae angustifoliae radix<br />
Echinaceae pallidae radix<br />
Echinaceae purpureae radix<br />
Equiseti herba<br />
Hamamelidis cortex<br />
Hamamelidis folium<br />
Hamamelidis folium et cortex,<br />
distillate<br />
Hippocastani semen<br />
Hyperici herba<br />
Lichen islandicus<br />
Meliloti herba<br />
Menthae piperitae folium<br />
Polygalae radix<br />
Polypodii radix<br />
Rhei radix<br />
Ribis nigri folium<br />
Rusci aculeati rhizoma<br />
Salicis cortex<br />
Salviae folium<br />
Sambuci flos<br />
Solidaginis virgaureae herba<br />
Thymi herba<br />
Urticae folium<br />
Urticae radix<br />
Uvae ursi folium<br />
Verbasci flos<br />
Zingiberis rhizome<br />
Assessment close to finalisation<br />
(pre-final)<br />
Anisi fructus<br />
Anisi aetheroleum<br />
Foeniculi amari fructus<br />
Foeniculi amari fructus aetheroleum<br />
Foeniculi dulcis fructus<br />
Draft published<br />
Echinaceae purpureae herba<br />
Passiflorae herba<br />
Primulae flos<br />
Primulae radix<br />
Rhamni purshianae cortex<br />
Draft under discussion<br />
Betulae folium<br />
Eleutherococci radix<br />
Harpagophyti radix<br />
Lupuli flores<br />
Melissae folium<br />
Menthae piperitae aetheroleum<br />
Urticae herba<br />
NML readers who want to know<br />
more can find it at:<br />
www.emea.eu.int/htms/human/<br />
hmpc/mpcmonographsadopt.htm/<br />
HORSE CHESTNUT<br />
RECOMMENDED BY<br />
WELLNESS LETTER ®<br />
The respected Wellness Letter ® ,<br />
published by the School of Public<br />
Health at the University of<br />
California at Berkeley, says horse<br />
chestnut extract might be worth<br />
trying for treatment of hemorrhoids<br />
since most over-the-counter<br />
treatments are of limited value.<br />
.<br />
See WELLNESS -- Cont’d on p. 9...<br />
www.NatMed<strong>Law</strong>.com<br />
to search back issues
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 9<br />
WELLNESS -- Cont’d from p. 8...<br />
The article in the May 2007 issue<br />
says that Aesculus hippocastanum<br />
has been in Europe for treatment<br />
of various vein disorders of the<br />
legs. In Germany they say the<br />
extract is approved for treatment<br />
of varicose veins and hemorrhoids.<br />
You can read the complete article<br />
at: www.wellnessletter.com/html/<br />
wl/wlAskExperts.html/<br />
In the 1998 First Edition of The<br />
Complete German E Commission<br />
Monographs/Therapeutic Guide to<br />
<strong>Herbal</strong> <strong>Medicine</strong>s by Blumenthal,<br />
Busse, Goldberg, Gruenwald, Hall,<br />
Klein, Riggins & Rister, the plant<br />
is also described as Hippocastani<br />
cortex/ -flos and its parts, the bark<br />
and flower are used as well as<br />
preparations thereof.<br />
Horse chestnut is combined with<br />
a large number of other herbs in<br />
these preparations. Commission E<br />
says there are no known risks, but<br />
since effectiveness is not<br />
documented, “a therapeutic<br />
application cannot be<br />
recommended.” p. 394. However,<br />
in the Approved Herbs section of<br />
the Commission E book there is<br />
listed Horse Chestnut seed which<br />
is a dry extract prepared from the<br />
seeds that is adjusted to 16 to 20<br />
percent triterpene glycodies<br />
(calculated as anhydrous aescin).<br />
Under the Approved Herb<br />
section clinical studies in humans<br />
are mentioned without citations of<br />
publications stating that with a<br />
placebo as reference, “a significant<br />
<strong>Natural</strong> <strong>Medicine</strong> Ties TM<br />
www.natmedlaw.com<br />
reduction of transcapillary<br />
filtration has been demonstrated in<br />
pharmacological studies involving<br />
human subjects, and a significant<br />
improvement shown in the<br />
symptoms of chronic venious<br />
insufficiency (sensation of<br />
tiredness, heaviness and tension,<br />
pruritus, pain and swelling in the<br />
legs) in various randomized<br />
double-blind studies and/or<br />
crossover studies.”<br />
The Approved <strong>Herbal</strong> monograph<br />
also states there are no known<br />
contraindications, special caution<br />
for use, or interaction with other<br />
drugs.<br />
It is important to remember that<br />
the German Commission E system<br />
which started in 1978 is be<br />
superceded by the European Union<br />
Adopted Community Monographs<br />
in a system that will require each<br />
member state to adopt the EU<br />
labeling over a period of time. At<br />
the present the only adopted EU<br />
monographs are for aloe, frangulae<br />
cortex, linum semen, plantaginis<br />
ovatae semen, plantaginis ovatae<br />
semen tegumentum, psyllii semen,<br />
senna fructus, senna folium, and<br />
valerianae radix. In plainer<br />
English these are aloe, frangula<br />
bark, linseed, plantago seed,<br />
plantago husk, psyllium seed,<br />
senna pods, senna leaf, and<br />
valerian root.<br />
As of March 20, 2007<br />
hippocastani semen has a<br />
“Rapporteur” assigned to the<br />
monograph process, which is the<br />
first stage of collecting data in the<br />
UE review system. See the<br />
complete list of in-process herbal<br />
drugs in the article on page 8. More<br />
information is at: www.emea.eu.int/<br />
pdfs/human/hmpc.<br />
NEW INGREDIENTS -- Con’d from p. 7...<br />
evidence indicates the product<br />
when used as suggested will reasonably<br />
be expected to be safe].<br />
Under 21 USC 350b(a)(2), there<br />
must be a history of use or other<br />
evidence of safety establishing that<br />
the new dietary ingredient, when<br />
used under the conditions recommended<br />
or suggested in the labeling<br />
of the dietary supplement, will<br />
reasonably be expected to be safe.<br />
If this requirement is not met, the<br />
dietary supplement is considered to<br />
be adulterated under 21 USC<br />
342(f)(1)(B) because there is inadequate<br />
information to provide a<br />
reasonable assurance that the new<br />
dietary ingredient does not present<br />
a significant or unreasonable risk<br />
of illness or injury. This letter is to<br />
alert you within the 75-day notification<br />
period that FDA intends to<br />
complete its evaluation within a<br />
few weeks and send you a response<br />
to your notification. Please note<br />
that a lack of a response to a notification<br />
within the 75-day<br />
timeframe does not constitute a<br />
finding by the agency that the ingredient<br />
or a product that contains<br />
the ingredient is safe or is not adulterated<br />
under 21 USC 342, See 21<br />
CFR 190.6(f).” [Because there is<br />
no other entry in this docket, and<br />
that multiple exchanges of information<br />
had taken place between<br />
Iwade and FDA before these documents<br />
were placed in the public<br />
file, it is presumed that this NDI<br />
was approved. Nevertheless,<br />
FDA’s preferred legalese in handling<br />
responses to these filings<br />
leaves matters confusing and unclear.<br />
– Ed.] Docket No. 1995S-<br />
0316, RPT 356, received at the<br />
Dockets Office on October 6, 2006,<br />
filed October 6, 2006, and posted<br />
See NEW INGREDIENTS -- Cont’d on p. 22...<br />
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside<br />
Page 10 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
EPHEDRA APPEAL -- Continued from page 1...<br />
Four’; that is, cert will be granted if a minimum of<br />
four of the nine justices favor the grant. This is not a<br />
written rule, but rather a long-standing tradition.<br />
Accordingly, cert has been denied even when as many<br />
as three justices have favored it. The philosophy is<br />
that if a ‘substantial minority’ feels the case should<br />
be heard and decided (not necessarily that it should<br />
be decided a certain way), the Court should consider<br />
the merits and decide the case.” 1 From 1980 to 2003<br />
thousands of Petitions are filed each year, but fewer<br />
than 100 are accepted. See chart from Judge<br />
Messitte’s article reproduced on page 1.<br />
Judge Messitte also says “Much can be said about<br />
what makes a petition worthy of a grant of cert or<br />
‘certworthy,’ as lawyers say. Perhaps the main point<br />
of interest is whether and how much the petitioner<br />
must argue the merits of his case in his petition.<br />
Inevitably, some part of the petition must do this, but,<br />
again, the primary showing must be whether there is<br />
a split of authority over the legal questions posed by<br />
the given case and/or why it is in the public interest<br />
that the questions be decided.” 2<br />
The Neutraceutical Petition, provided courtesy of<br />
its counsel, Emord & Associates, to NML gives<br />
several good arguments and four Justices may want<br />
to send FDA a message about statutory interpretation.<br />
Here are some of the arguments being made by the<br />
Petitioners.<br />
The major statutory construction argument is all<br />
about what “or” means. In setting up the arguments,<br />
the Petition states: “Although FDA refused to permit any<br />
exception to its ban on dietary supplements, it did exclude<br />
from its ban all ephedrine alkaloid containing foods as<br />
well as all traditional OTC Asian medicines (regardless of<br />
ephedrine alkaloid dose). Therefore, it limited its ban to<br />
dietary supplements. See App. 70-73. The FDA decision<br />
thus produced the illogical and irrational result of removing<br />
ephedrine alkaloids at every quantitative amount from the<br />
market when in dietary supplements but of not removing<br />
ephedrine alkaloids in any quantitative amount from the<br />
market when in other foods. The Final Rule thus prohibited<br />
petitioners from selling their raw crushed Ephedra sinica<br />
herb in a gelatin capsule (a dietary supplement form) but<br />
did not prohibit them from selling the very same substance<br />
as a tea (a food form). The FDA decision thus defines<br />
adulteration law in a way that discriminates based on the<br />
form of dietary ingredient rather than on the substance of<br />
the ingredient, such that dietary ingredients sold as foods<br />
or as traditional OTC Asian medicines enjoy a statutorily<br />
protected position<br />
denied dietary<br />
ingredients sold<br />
as dietary<br />
supplements.” 3<br />
“To reach this<br />
odd result, the<br />
FDA had to adopt<br />
a novel standard<br />
for proving<br />
ephedrine<br />
alkaloids harmful,<br />
a standard<br />
applicable to all<br />
d i e t a r y<br />
supplements and<br />
in conflict with the<br />
pre-existing<br />
NPA AMICUS BRIEF<br />
The <strong>Natural</strong> Products Association<br />
(formerly the National Nutritional<br />
Foods Association)<br />
filed an Amicus Curiae brief<br />
with SCOTUS on April 6, 2007<br />
in the Neutraceutical International<br />
case concerning the ephedra<br />
ban. NPA represents over<br />
10,000 manufacturers, wholesalers,<br />
distributors and retailers<br />
of natural products, including<br />
dietary supplements.<br />
See AMICUS --Continued on page 19...<br />
standard for foods, including dietary supplements. The<br />
standard, adopted for all dietary supplements, not just<br />
EDS, fundamentally alters adulteration law for those<br />
products….”<br />
In FDA’s final rule, the agency declared dietary<br />
supplements adulterated if the contained so much as a<br />
single molecule of ephedrine alkaloids and did not rely on<br />
any evidence of the levels where toxicity occurred. Thus<br />
FDA abandoned the Paraselsian axiom that is the basis<br />
of all toxicology – “[a]ll things are poison and nothing is<br />
without poison. Solely the dose determines that a thing is<br />
not a poison.” This position also abandons the basis<br />
for the law of adulteration, the Petition said.<br />
What FDA did was to adopt a new adulteration<br />
test “articulating a standard for unreasonable risk under<br />
402(f) (1) (A) of the [FDCA] for the first time…. ‘In so<br />
doing it took the interpretive leap of equating the<br />
‘unreasonable risk’ statutorily prescribed for dietary<br />
supplements with the ‘safe and effective’ standard that<br />
the FDCA uses to assess whether new drugs may be<br />
granted approval to enter the market. See FDA, 21 U.S.C.<br />
§ 355(b)(1)(A); see also Guidance for Industry: Premarket<br />
Risk Assessment, 5, n.4 (2005) (associating FDA’s risks<br />
and benefits’ test with statutory requirements that drugs<br />
be safe and effective). Thereafter, without specific statutory<br />
authorization, FDA adopted the following three-step<br />
approach.”<br />
“Step 1. The FDA read the statutory words<br />
‘unreasonable risk’ to permit the FDA to use a generalized<br />
risk/benefit standard to determine whether a dietary<br />
supplement should remain on the market. App. 75, 105-<br />
116.”<br />
“Step 2. The FDA assigned minimal health benefits to<br />
ordinary uses, including important consumer objectives<br />
See EPHEDRA APPEAL -- Continued on page 11...
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 11<br />
STRATEGY -- Continued from page 6...<br />
2. Completed Product License Application<br />
Form (PLA-FORM);<br />
3. Copy of the label most recently approved by<br />
the TPD;<br />
• Copy of product information printed from<br />
the TPD Drug Products Data base (DPD).<br />
More information is available at: www.hc-sc.gc.ca/<br />
dhp-mps/prodnatur/bulletins/din_trans_din_e.html<br />
4. Copy of the proposed product label text<br />
meeting the labelsing requirements for NHPs<br />
Sections 93 - 95 of the <strong>Natural</strong> Health<br />
Products <strong>Regulation</strong>s. Also refer to Chapter 3<br />
of the Labeling Guidance Document).<br />
5.<br />
NOTE: if the only claim on the TPD label is<br />
“For therapeutic use only”, applications for<br />
vitamin and mineral products must include the<br />
health claim(s) indicated on the applicable<br />
NHPD monograph(s) (see: Compendium of<br />
Monographs). Risk statements from<br />
applicable NHPD monographs should be<br />
added to the label;<br />
6. Copy of the finished product specifications<br />
including identity, purity and potency;<br />
NOTE: if it is unclear as to which tests are<br />
used to identify the medicinal ingredients in<br />
the finished product, the raw material<br />
specification for that medicinal ingredient<br />
should be included as well;<br />
7. Completed animal tissue forms, if applicable.<br />
NOTE: Specified risk materials (SRMs) are<br />
prohibited, therefore, if any non-medicinal<br />
ingredients are SRMs they must be removed or<br />
replaced by a non-SRM source material. This must<br />
be done in order to be categorized as a Transitional<br />
DIN Product Application; and Product registration<br />
information:<br />
• Copy of Drug Notification form or Notice<br />
of Compliance<br />
www.natmedlaw.com<br />
EPHEDRA APPEAL -- Continued from page 10...<br />
such as weight loss, enhanced athletic performance,<br />
increased energy levels, easier breathing, and other uses,<br />
finding among them only weight loss sufficient to warrant<br />
critical assessment. FDA then determined that scientific<br />
evidence concerning those effects assessed ‘short term’<br />
rather than ‘long term’ weight loss, and ‘short term’ weight<br />
loss was not a significant health benefit according to FDA.<br />
App. 119-121.<br />
“Step 3. The FDA hired Dr. Mario Inchiosa to evaluate<br />
the scientific literature on the effects of intravenously<br />
injected drugs epinephrine and ephedrine, effects it<br />
claimed would occur in orally ingested EDS. Inchiosa’s<br />
untested assessment, in the form of letters of opinion,<br />
concluded that no clear evidence established that the<br />
drugs epinephrine and ephedrine were safe at any<br />
dosages. Dr. Inchiosa created his own unique unpeerreviewed<br />
and untested hypothetical extrapolation in which<br />
he presumed EDS would affect the body in a manner<br />
chemically indistinguishable from ephedrine and<br />
epinephrine, both potent drugs. He extrapolated from the<br />
studies involving intravenous injection of those drugs, even<br />
though the dietary ingredient is orally ingested. He<br />
assumed comparable effects, despite the fact that the<br />
drugs are very potent inducers of rapid heart contractions<br />
and elevated blood pressure and directly affect parts of<br />
the body that the dietary ingredient does not. He employed<br />
a mathematical model never before used in pharmacology<br />
or toxicology to compare the drugs with the dietary<br />
ingredient. Based on those unconventional and<br />
unprecedented techniques, he concluded that continuous<br />
ingestion of 1.5 mg of ephedrine alkaloids every four hours<br />
would present a risk of injury comparable to the drugs.<br />
Neither Dr. Inchiosa nor anyone else at FDA ever<br />
discussed, let alone evaluated clinically, ingestion of 5 mg<br />
or less of ephedrine alkaloids twice daily at meal times,<br />
the actual dosing in the conditions of use for the petitioners’<br />
product. Dr. Inchiosa’s letters were not peer-reviewed.<br />
His hypothesis was never tested. His conclusions of<br />
toxicity were not based on any clinical testing of the actual<br />
dietary ingredient at any dose level, let alone 10 or fewer<br />
mg. The drug-dietary ingredient comparison model he<br />
invented was not adopted through scientific consensus<br />
and is not a standard method for pharmacological or<br />
toxicological evaluation, yet that comparison acquired<br />
decisional significance in the Final Rule’s assessment of<br />
low dose ephedra. App. 101.”<br />
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside<br />
See EPHEDRA APPEAL -- Continued on page 12...
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside<br />
Page 12 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
Photo Courtesy of Lloyd Library and Museum<br />
EPHEDRA APPEAL -- Continued from page 11...<br />
“On the strength of this record, the<br />
FDA concluded that it had carried its<br />
burden of proof that ephedrine<br />
alkaloids posed a risk of illness or<br />
injury at every dose level, including<br />
daily dose amounts of 10 mg or less,<br />
justifying a total ban. In words that<br />
track the statute:<br />
The [FDA] is issuing a final<br />
regulation declaring dietary<br />
supplements containing<br />
ephedrine alkaloids adulterated<br />
under the [FDCA] because<br />
they present an unreasonable<br />
risk of illness or injury<br />
under the conditions of<br />
use recommended or suggested<br />
in labeling, or if no<br />
conditions of use are suggested<br />
or recommended in<br />
labeling, under ordinary conditions<br />
of use.<br />
App. 46.<br />
“As should be apparent from the<br />
FDA’s calculated omission of the word<br />
‘significant’ in the quoted passage,<br />
the FDA did not find that ephedrine<br />
alkaloids posed a ‘significant risk’ of<br />
harm. Its ruling is therefore wholly<br />
consistent with the proposition that<br />
ephedrine alkaloids may present at 10<br />
mg or less per daily dose amounts<br />
only a miniscule risk of harm in their<br />
specified use. Stressing the word ‘or’<br />
as disjunctive in the phrase ‘significant<br />
or unreasonable risk’ (and ignoring<br />
‘significant risk’ entirely), the FDA<br />
concluded solely that the risk,<br />
however small, was unreasonable in<br />
light of the minimal health benefits it<br />
stated the substance provided. FDA<br />
therefore concluded that it was wholly<br />
‘unnecessary’ to ask whether the risk<br />
was significant because the term was<br />
included in the statute as a strict<br />
alternative to the phrase<br />
‘unreasonable risk.’ App. 70 (‘We are<br />
not addressing the issue of whether<br />
these products present a ‘significant’<br />
risk under section 402(f)(1)(A) of the<br />
act’); see also Def. Mem. Supp.<br />
Cross-Mot. Summ. J. at 31 n.14<br />
(‘Because the FDA concluded that<br />
EDS pose an unreasonable risk, it<br />
was not necessary for the agency to<br />
address DSHEA’s significant risk<br />
standard’). In sum, the FDA’s position<br />
is that ephedrine alkaloids present an<br />
insignificant, but unreasonable risk of<br />
illness or injury at 10 mg or less (but<br />
no significant health benefit at any<br />
dose).”<br />
Further along in the Petition,<br />
Nutraceutical says that the 10 th<br />
Circuit held the term<br />
“unreasonable risk” required the<br />
risk-benefit comparison as<br />
prescribed in the New Drugs<br />
section of the FDCA. And the 10 th<br />
Circuit violated the Supreme<br />
Court’s holding that reading<br />
wording in a statute, in this case<br />
“unreasonable risk” in isolation<br />
from all other terms in the statute<br />
to avoid treating the words as<br />
surplusage is unacceptable in when<br />
used to interpret the law contrary<br />
to congressional intent. Scheidler<br />
v. National Organization for<br />
Women, Inc. 126 S. Ct. 1264, 1273<br />
(2006)<br />
The Petition then argues that “But<br />
even under the surplusage canon, the<br />
Tenth Circuit’s decision fails because<br />
it has not satisfied the statutory<br />
requirement that adulteration be<br />
determined based on recommended<br />
or suggested dosages in labeling. The<br />
surplusage canon hardly requires the<br />
FDA to introduce through the back<br />
door a fundamental regime change<br />
whereby dietary supplements are now<br />
evaluated under the standards<br />
reserved for drugs in the FDCA with<br />
the FDA bearing (an attenuated)<br />
burden of proof.”<br />
“The Tenth Circuit has ruled out the<br />
possibility of a simpler reading, one<br />
consistent with the plain language of<br />
the statute and the legislative intent.<br />
‘Unreasonable’ need not beget a<br />
meaning independent of all other<br />
words in the statutory sentence nor<br />
does it require consideration of health<br />
benefit when no reference to a<br />
comparison of risk with benefit exists<br />
in the Adulterated Food section or in<br />
the legislative history pertaining to that<br />
section. ‘Unreasonable’ does not<br />
require abandonment of the central<br />
principle of adulteration law, the<br />
Paracelsian axiom that dose<br />
determines toxicity. Within the<br />
meaning of the statute, a risk<br />
becomes ‘unreasonable’ at a dose<br />
level that causes illness or injury. A<br />
risk becomes ‘significant’ based on<br />
the degree of illness or injury<br />
experienced (e.g., a tumor as<br />
opposed to a headache). The term ‘or’<br />
does not carry a disjunctive meaning,<br />
such that FDA is at liberty to ignore<br />
‘significant’ risk in finding adulteration.<br />
Rather, each term independently<br />
modifies the noun ‘risk’ and must be<br />
interpreted. Harmonized with the food<br />
subparts of Section 342, a ‘significant<br />
or unreasonable risk’ is one that<br />
causes illness or injury at a particular<br />
dose level (and above) that cannot be<br />
mitigated through modification of<br />
labeling but is a characteristic<br />
response experienced by users to the<br />
dose recommended, suggested, or<br />
ordinarily consumed. Moreover,<br />
‘unreasonable’ in the dietary<br />
ingredient subsection of the<br />
Adulterated Food section does not<br />
invite a construction of the FDCA that<br />
supports the bizarre distinction that<br />
defines a food containing 15 to 30 mg<br />
See EPHEDRA APPEAL --Cont’d on p.13...
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 13<br />
EPHEDRA APPEAL -- Continued from p. 12...<br />
or more of a dietary ingredient exempt<br />
from the holding of adulteration but<br />
declares a dietary supplement<br />
containing 10 mg or less of the same<br />
dietary ingredient adulterated and<br />
banned from the market. Congress<br />
intended that the dietary ingredient<br />
subsection be harmonized with the<br />
pre-existing statute, S. Rep. No. 103-<br />
410 at 21, not interpreted to create a<br />
separate regime in conflict with the<br />
old. The FDA’s view upheld by the<br />
Tenth Circuit is clearly incorrect<br />
because it exempts all ephedrine<br />
alkaloid-containing foods, including<br />
ephedra tea (having ephedrine<br />
alkaloids per serving in concentrations<br />
greater by far than the 10 mg per daily<br />
dose in the petitioners’ dietary<br />
supplements). Indeed, the herb used<br />
in ephedra tea is precisely the same<br />
as the herb used in Petitioners’ dietary<br />
supplement (raw crushed ephedra<br />
sinica), differing only in the amount of<br />
ephedrine alkaloids delivered (15 to<br />
30 mg or more from the tea per<br />
serving compared to 10 mg or less<br />
mg from the capsule per daily dose).<br />
DSHEA contains no intimation that<br />
Congress has authorized an absurd<br />
reading of the Adulterated Food<br />
section, one that construes a single,<br />
uniform statutory section to yield an<br />
internally inconsistent result, against<br />
the statutory construction<br />
requirements of this Court. See FDA<br />
v. Brown & Williamson Tobacco Corp.,<br />
529 U.S. 120, 133 (2000).”<br />
The Petition next says FDA has<br />
fundamentally altered food<br />
adulteration law without<br />
authorization from Congress and<br />
the 10 th Circuit has condoned this<br />
usurpation of legislative power.<br />
FDA relied on a non sequitur: “that<br />
the absence of evidence proving<br />
safety is the same as the presence<br />
of evidence proving harm. Quite<br />
simply, FDA has smuggled a<br />
finding of significant risk into its<br />
unbalanced risk-benefit analysis.”<br />
And finally, the Petition<br />
summarizes: “In sum, if the FDA is<br />
allowed to conduct future analyses of<br />
dietary supplements under the<br />
skewed test that it has adopted and<br />
the Tenth Circuit has upheld, then the<br />
congressional compromise that<br />
defines separate regimes for foods<br />
and drugs no longer holds true in the<br />
food adulteration context. Rather, the<br />
FDA may henceforth draw inferences<br />
about the risks of dietary supplements<br />
at any dose level whatsoever, in the<br />
face of the statutory requirement that<br />
it run its evaluation at the<br />
recommended or suggested levels of<br />
use. It may henceforth evade its<br />
burden of proof by relying on unpeerreviewed,<br />
solicited opinion letters that<br />
present untested models that<br />
extrapolate from results of tests<br />
conducted on different substances<br />
(drugs) intravenously injected and<br />
presume those results the same for<br />
dietary ingredients orally ingested.<br />
Under the Tenth Circuit’s decision,<br />
FDA may now insist that infinitesimal<br />
risks are unreasonable, magnifying<br />
them beyond the record evidence, as<br />
it has done in Nutraceutical’s case,<br />
and then conduct a statutorily<br />
unauthorized benefit comparison that<br />
ignores all non-health benefits and<br />
subjectively deems virtually all health<br />
benefits insufficiently weighty to<br />
counterbalance perceived risks.<br />
Indeed, significant and clinically<br />
proven health benefits, presumably<br />
the kind that might be viewed by FDA<br />
as sufficiently weighty, cannot be<br />
demonstrated because intent to<br />
produce therapeutic benefits defines<br />
a drug and is impermissible for<br />
supplements.”<br />
“In short, the FDA has nullified the<br />
bedrock of adulteration law, that dose<br />
determines toxicity. It has negated the<br />
fundamental requirement that FDA<br />
prove a dietary ingredient itself, rather<br />
than a surrogate substance, i.e., a<br />
drug, a significant risk before banning<br />
the ingredient from the market.<br />
Without statutory authorization and<br />
against the plain and intended<br />
meaning of Congress, FDA has<br />
superimposed on the Adulterated<br />
Food section a drug risk-benefit<br />
standard, eliminating the essential<br />
divide between foods and drugs<br />
created by the FDCA and rendering<br />
the critically important Adulterated<br />
Food section internally inconsistent<br />
and irrational. That rewrite of the law<br />
is causing instability in food and<br />
supplement markets. <strong>Law</strong>less<br />
behavior by agencies in defiance of<br />
their enabling statutes should never<br />
be tolerated when the means are at<br />
hand to correct that behavior.”<br />
Imagine reading several<br />
thousands of these petitions and<br />
trying to sift out fewer than 100 to<br />
ask counsel for more briefs and<br />
argument. The job will take<br />
concentration and maybe some<br />
enthusiasm, but issues about<br />
government agencies writing their<br />
own laws have always been of<br />
interest to Supreme Court Justices.<br />
If the Court ignores such rogue<br />
government overreaching as<br />
happened in this case, then<br />
Congress is likely to react.<br />
The Petition for Certiorari is 29<br />
pages and the Court’s rule limits it to<br />
30 pages. There is an appendix of 137<br />
pages. Richard A. Epstein, Esq., who<br />
is the James Parker Hall<br />
Distinguished Service Professor of<br />
<strong>Law</strong>, Faculty Director for Curriculum,<br />
and Director, <strong>Law</strong> and Economics<br />
Program at the University of Chicago,<br />
(773) 702-9563) is on the Petition<br />
with Jonathan W. Emord, Esq. of<br />
Reston, Virginia, (202) 466-6937)<br />
who is the Counsel of Record.<br />
(Footnotes)<br />
1 eJournal USA: Issues of Democracy,<br />
April 2005, http://usinfo.state.gov/<br />
journals/itdhr/0405/ijde/messitte.htm<br />
2 Ibid.<br />
3 All additional quotations are from the<br />
Petition for Certiorari.<br />
Nutraceutical Corporation; Solaray, Inc.<br />
v. Andrew Von Eschenbach,<br />
Commissioner, et al., United States<br />
Supreme Court, No. 06-922.<br />
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside<br />
Page 14 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
SECTION 403 LETTERS - Continued from page 4...<br />
Holistic Therapies of Boca Raton, Florida, wrote<br />
FDA on November 8, 2006 stating that it was a<br />
distributor for the product MegaBone, made by<br />
Orthomolecular Products of Stevens Point,<br />
Wisconsin. MegaBone contains Vitamins D3, K,<br />
Folic Acid, Calcium, Phosphorus, Magnesium,<br />
Selenium, Copper, Manganese, Molybdenum,<br />
Ipriflavone, Strontium Citrate, Montmorillionite,<br />
Boron, Cranberry Juice Concentrate and Tumeric<br />
Root Extract. There were no claims and the letter<br />
was unsigned over the words “[Your name here}.”<br />
There was one other notice letter included, also<br />
unsigned, but this time with a copy of the label for<br />
the product ThyroMega, containing Iodine, L-<br />
Thyrosine USP, Ashwaganda Root Extract,<br />
Bladderwrack Leaf and again no claims. FDA’s<br />
Vasilios H. Frankos, Ph.D. responded to the<br />
“President” at the company address by letter dated<br />
November 28, 2006 and stated that since the notices<br />
submitted were not signed they do not meet the<br />
requirements of 21 CFR 101.93(a)(3) and as such do<br />
not comply with the Act. Docket No. 97S-0163, Ltr<br />
913, received at the Dockets Office on December 18,<br />
2006, entered into the Docket on December 22, 2006,<br />
and posted to the FDA Website on January 10, 2007.<br />
Tronex Company of Parsippany, New Jersey,<br />
wrote FDA on October 31, 2006 to give notice that it<br />
represented Wellslife Corporation of Taiwan as its<br />
authorized U.S. Agent and Principal Distributor for<br />
products named Eydeal, Soligen, Beauty Sleep,<br />
Trimnastics and Trimnastics Tea, CD – Cup D’Jour,<br />
Celline, and Hair 3 System. However, FDA included<br />
in the public file information pages on only two of<br />
the eight products. [NML has requested FDA to post<br />
to the website the other eight pages. – Ed] The page<br />
on Trimnastics Tea indicates it contains Rose, Flower<br />
Bud of Seville orange, Jasmine, Chunxiong Rhizome,<br />
and Lotus Leaf. Trimnastics Tea is to be used one<br />
teach a time and two times a day in hot water to steep<br />
2 to 4 minutes. There are 60 tea bags in a box. The<br />
claim statement offered is: “Trimnastics Tea is the<br />
safe and natural companion to Trimnastic. It<br />
compliments the daily regime by reminding you to<br />
take a weight-reduction tea break. Trimnastics and<br />
the “TEA” are made from all natural herbal<br />
ingredients.” Beautysleep is recommended for use<br />
by taking one packet per day with warm water one<br />
hour before bedtime. There are 30 packets in a box.<br />
The contents of a packet are Zizphus 800 mg, Oyster<br />
shell 600 mg, Rehmanniae 600 mg, Spica Pruneellae<br />
500 mg, and White Peony Root 500 mg. The claim<br />
statement for Beautysleep is: “Beauty Sleep(sic) helps<br />
you enjoy good sleep naturally without taking the risk<br />
of prescription drugs dependency. Beauty Sleep(sic)<br />
is based on a Traditional Chinese <strong>Medicine</strong> formula,<br />
and made from all natural herbal ingredients. It is<br />
absolutely safe and effective.” FDA responded to<br />
Peter Sewell by letter of November 28, 2006 signed<br />
by Vasilios H. Frankos, Ph.D. to these two notices.<br />
Frankos wrote that Trimnastics Tea, as a drink, is a<br />
conventional food and must comply with nutrition<br />
labeling defined at 21 CFR 101.9. Any ingredient in<br />
food must be GRAS or an approved food additive or<br />
the food is adulterated and cannot be legally marketed<br />
under the Act. FDA wrote that the claims for<br />
Beautysleep “Helps you fall asleep if you have<br />
difficulty falling asleep” and “helps to reduce difficulty<br />
falling asleep” are disease claims. [Neither of these<br />
claims are in the documents FDA placed in the public<br />
file. – Ed.] Also, FDA stated that the January 6, 2000<br />
rule on structure/function claims does not permit a<br />
dietary supplement manufacturer to state that it has<br />
fewer side effects than a drug. Because of these claims<br />
Beautysleep will be regulated as a drug if the claims<br />
are used. Docket No. 97S-0163, Ltr 914, received at<br />
the Dockets Office on December 18, 2006, entered<br />
into the Docket on December 22, 2006, and posted to<br />
the FDA Website on January 10, 2007.<br />
Alert Nutrition, LLC of Wayzata, Minnesota wrote<br />
FDA on November 11, 2006 to give three notices that<br />
it would use certain “label revision” statements of<br />
claim on its products, Immune Alert R Children’s<br />
Formula – Cherry Flavor Chewable Tablets, Immune<br />
Alert R Adult’s Formula – Orange Flavor Chewable<br />
Tablets, and Immune Alert R Adult’s Formula –<br />
Capsules. Each letter was accompanied by a product<br />
label and a copy of the cover of a pull out Spanish<br />
label and cap sticker. For Immune Alert R Children’s<br />
Formula – Cherry Flavor Chewable Tablets contain<br />
BetaRight R of beta 1,3/1,6 glucan extracted from foodgrade<br />
Baker’s yeast (Saccharomuyces cerevisiae), a<br />
FDA-approved ingredient (21 CFR 170.30) and the<br />
claim was “Germs, virus, bacteria … kids pass them<br />
back and forth and bring them home to you. Taken<br />
daily, Immune Alertwith Beta 1,3/1,6 Glucan (an all<br />
See SECTION 403 LETTERS -- Continued on page 15... .
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 15<br />
SECTION 403 LETTERS -- Continued from page 14...<br />
natural complex carbohydrate extracted from baker’s<br />
yeast) puts your children’s natural defenses on high<br />
alert to fight off these invaders.” FDA’s December<br />
8, 2006 letter from Vasilios H. Frankos, Ph.D. to<br />
Jerome Licari, Ph.D. stated that this claim was a<br />
disease claim, namely diseases caused by pathological<br />
organisms. For Immune Alert R Adult’s Formula –<br />
Orange Flavor Chewable Tablets, and Immune Alert R<br />
Adult’s Formula – Capsules, containing the same<br />
ingredients, the claims also were the same, namely,<br />
“Each day your body fights off billions of diseasecausing<br />
micro-organisms. Taken daily, Immune<br />
Alertwith Beta 1,3/1,6 Glucan (an all natural complex<br />
carbohydrate extracted from baker’s yeast) puts your<br />
children’s natural defenses on high alert to fight off<br />
these invaders – stopping them before they stop you.”<br />
FDA’s letter also stated that these claims were disease<br />
claims and if Alert Nutrition intends to make these<br />
claims, the products are subject to regulation as drugs<br />
under the Act. Docket No. 97S-0163, Ltr 915,<br />
received at the Dockets Office on December 18, 2006,<br />
entered into the Docket on December 22, 2006, and<br />
posted to the FDA Website on January 10, 2007.<br />
Enzymatic Therapy, Inc. of Green Bay, Wisconsin,<br />
filed a notice, dated November 10, 2006 to the FDA<br />
for the product LeveLife Synergy Drink containing<br />
taurine, Benco Synergy 1, whey protein,<br />
Glucomannan Extract, Chromium picolinate, Malic<br />
Acid, Hersperidin Complex 50%, Vitamin C, L-<br />
Tyrosine, Glycine, Roboflavin, and Inositol. The<br />
amounts are not disclosed in the filing. There are<br />
thirteen claims made for the product. FDA’s Vasilios<br />
H. Frankos, Ph.D. responded to Michael P. Devereaux<br />
by letter of December 8, 2006 stating that a drink is a<br />
conventional food and not a dietary supplement.<br />
Therefore the product should be labeled with nutrition<br />
labeling and all ingredients must be GRAS or<br />
approved food additives or the product is adulterated<br />
and may not be marketed in the United States. Docket<br />
No. 97S-0163, Ltr 916, received at the Dockets Office<br />
on December 18, 2006, entered into the Docket on<br />
December 22, 2006, and posted to the FDA Website<br />
on January 10, 2007.<br />
Calgenex Corporation of Tampa, Florida, filed a<br />
notice, dated November 29, 2006 for its product<br />
Omeganol, containing fish oil concentrate, d-alpha<br />
tocotrienol, d-beta tocotrienol, d-gamma tocotrienol,<br />
d-delta tocotrienol and d-alpha tocopherol. The<br />
ingredient amounts were not indicated. The claims<br />
were: “Manage your cholesterol and triglycerides.<br />
A natural solution to help maintain blood lipid<br />
balance. Helps protect your heart.” FDA’s Vasilios<br />
H. Frankos, Ph.D. responded to Grant Carlson by<br />
letter of December 13, 2007, stating that the preamble<br />
to the final rule on structure/function claims, January<br />
6, 2000, explained that claims about maintenance of<br />
normal cholesterol levels do not necessarily constitute<br />
implied disease claims, but the claims would have to<br />
clarify that the product is only for maintenance of<br />
cholesterol levels that are already within the normal<br />
range. Without such an addition, the claims being<br />
made are disease claims making the product subject<br />
to regulation as a drug under the Act. Docket No.<br />
97S-0163, Ltr 917, received at the Dockets Office on<br />
December 18, 2006, entered into the Docket on<br />
December 22, 2006, and posted to the FDA Website<br />
on January 10, 2007.<br />
World Nutrition, Inc. of Scottsdale, Arizona,<br />
wrote FDA on December 11, 2006 to give notice that<br />
See SECTION 403 LETTERS -- Continued on page 16...<br />
PHOTOGRAPHS FROM LLOYD LIBRARY AND MUSEUM<br />
Cover -- Common Horse Chestnut (æsculus hippocanstnum) from<br />
MEDICAL BOTANY by John Stephenson and James Morss Churchill,<br />
M.D. 1829.<br />
Cover -- Podophyllum (Podophyllum Peltatum) from AMERICAN<br />
MEDICINAL PLANTS by Charles F. Millspaugh, M.D, 1887<br />
Cover --Goldenseal (Hydrastis Canadensis) from AMERICAN ME-<br />
DICINAL PLANTS by Charles F. Millspaugh, M.D, 1887<br />
Page 4 - Foxglove (Digitalis purpurea) from MEDICAL BOTANY<br />
by John Stephenson, M.D. and James Morss Churchill, F.L.S.<br />
1828<br />
Page 12 - Ginseng (Panax quinquefolium) from THE FAMILY FLORA<br />
AND MATERIA MEDICA BOTANICA by Peter P. Good 1847<br />
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside<br />
www.natmedlaw.com<br />
Page 16 - Olive Tree (Olea Europæa) from FLORA GRÆCA by<br />
Johannes Sibthorp. M.D. & Jacobus Edvardus Smith, M.D., 1806<br />
Page 22 -- Cone Flower (Echinacea Intermedia) from PAXTON’S<br />
MAGAZINE OF BOTANY, 15th Vol. 1849
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside<br />
Page 16 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
SECTION 403 -- Contintued from p. 15..<br />
it would use a certain claim for its<br />
product, Wang’s Juemingjing Herb<br />
Extract, containing Flatstem,<br />
Milkvetch Seed, Chinese Wild<br />
Yam, Safflower, Milkvetch root,<br />
Cape Jasmine fruit, Kudzu vine<br />
root, Licorice Root, Korean<br />
ginseng. The amounts of<br />
ingredients are not disclosed. FDA<br />
wrote Tom Miano on December 13,<br />
2006 that the notice must be<br />
certified by a responsible<br />
individual that the notice is<br />
complete and accurate and that the<br />
notifying firm has substantiation<br />
that the statement is truthful and not<br />
misleading. FDA’s letter signed by<br />
Vasilios H. Frankos, Ph.D. did not<br />
comment further. Docket No. 97S-<br />
0163, Ltr 918, received at the<br />
Dockets Office on December 18,<br />
2006, entered into the Docket on<br />
December 22, 2006, and posted to<br />
the FDA Website on January 10,<br />
2007.<br />
Alltech Biotechnology Center<br />
of Nicholasville, Kentucky, sent<br />
FDA a letter on December 5, 2006<br />
to give notice that it would make<br />
statements of nutritional support<br />
for its product SEL-PLEX –<br />
mineral supplement – capsule. The<br />
ingredients of the product are<br />
selenium yeast, magnesium state an<br />
gelatin. The claims were:<br />
“Mineral supplement to maintain<br />
health. Selenium may reduce the<br />
risk of certain cancers. Some<br />
scientific evidence suggests that<br />
consumption of selenium may<br />
reduce the risk of certain forms of<br />
cancer, However, FDA has<br />
determined that this evidence is<br />
limited and not conclusive.<br />
Selenium may produce<br />
anticarcinogenic effects in the<br />
body. Some scientific evidence<br />
suggests that the consumption of<br />
selenium may produce<br />
anticarcinogenic effects in the<br />
body. However, FDA has<br />
determined that this evidence is<br />
limited and not conclusive.” FDA<br />
responded to Pearce Lyons, Ph.D.<br />
by letter, dated December 22, 2006<br />
from Vasilios H. Frankos, Ph.D.<br />
stating that this claim is not a<br />
structure/function claim, but a<br />
claim under 21 USC 343(r)(1)(B)<br />
because it represents the product<br />
will reduce the risk of a disease.<br />
FDA explained that it had<br />
previously agreed to exercise<br />
enforcement discretion for<br />
selenium products and claims for<br />
selenium at www.cfsan.fda.gov/<br />
~dms/ds-ltr35.htm and failure to<br />
make a claim in accordance with<br />
the enforcement discretion would<br />
cause the product to be<br />
misbranded. FDA added that using<br />
the proposed claim would result in<br />
the product being regulated as a<br />
drug because it claims the product<br />
is used to treat cancer, a disease.<br />
Docket No. 97S-0163, Ltr 919,<br />
March 1, 2007, entered into the<br />
Docket on February (sic) 1, 2007,<br />
and posted to the FDA Website on<br />
March 29, 2007.<br />
New Chapter, Inc. of<br />
Brattleboro, Vermont, wrote FDA<br />
on December 13, 2006 to give<br />
notice that it would use certain<br />
claims for its products<br />
Rhodiolaforce 100 and<br />
Rhodiolaforce 300. The product<br />
contains Rhodiola rosea. The<br />
claims were the same for these<br />
products, and were: (1) Thriving in<br />
high altitudes and in nature’s most<br />
challenging climates, this most<br />
resilient herbal is intensively<br />
studied for calming stress,<br />
increasing energy, metabolism of<br />
fat, enhancing memory, and<br />
boosting the body’s immune<br />
Olive Tree (Olea Europæa) 1806<br />
Photo Courtesy of Lloyd Library and Museum<br />
defenses. 2) Rhodiola also protects<br />
the heart in several ways, including<br />
boosting the energy available to<br />
heart muscles, thereby preventing<br />
the kind of damage that can lead<br />
to heart attacks or heart failure. 3)<br />
And its unique group of antioxidant<br />
compounds also helps to protect<br />
cells and DNA from being<br />
damaged by unstable oxygen<br />
compounds.” FDA responded to<br />
Ashley Chapin by letter of January<br />
3, 2007 from Vasilios H. Frankos,<br />
Ph.D. stating that the claim<br />
“Rhodiola also protects the heart<br />
… preventing the kind of damage<br />
that can lead to heart attacks or<br />
heart failure” was a disease claim<br />
for treating heart disease and stated<br />
that if this claim is used, the<br />
products are subject to regulation<br />
as a drug under the Act. Docket<br />
No. 97S-0163, Ltr 920, received at<br />
the Dockets Office on March 1,<br />
2007, entered into the Docket on<br />
February (sic) 1, 2007, and posted<br />
See SECTION 403 LETTERS -- Cont’d p. 17...
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 17<br />
SECTION 403 -- Continued from p.16 ...<br />
to the FDA Website on March 29,<br />
2007.<br />
Global Pharmatech, Inc. of<br />
Gaithersburg, Maryland wrote<br />
FDA on December 13, 2006 to give<br />
notice of its intent to distribute the<br />
product Vigor Supplier ® Tablet,<br />
containing Echinacea purpurea<br />
extract 0.4 g and American ginseng<br />
extract 0.1 g, made by Jilin<br />
Bencaotang Pharmceutical Co.,<br />
Ltd of China. The company also<br />
apparently wrote on December 12,<br />
2006 to submit a notice for the<br />
product Radish Seed Tablets,<br />
containing 1.4 g of Radish seed<br />
extract. FDA’s letter in response,<br />
dated January 16, 2007, signed by<br />
Vasilios H. Frankos, Ph.D. to Tom<br />
Du, M.D., Ph.D. states that the<br />
label for Vigor Supplier ® Tablet<br />
bears the statement “Echinacea<br />
has been used for the enhancement<br />
of human health for centuries.<br />
Anti-fatigue activity of American<br />
ginseng is well recognized in<br />
American and Asian community<br />
(Integr Cancer Ther. 2003 Sep:<br />
2(3):247.” The label for Radish<br />
Seed Tablets bears the statement<br />
“Chinese scientist observed that<br />
Radish seed extract is helpful to<br />
maintain human blood pressure in<br />
normal range. Pakistan scientists<br />
found that extract of Radish Seeds<br />
was able to decrease blood<br />
pressure in experimental animals<br />
(Fundam Clin Pharmacol 2006<br />
Feb; 20(1)57,” and “Chinese<br />
herbalists also showed that Radish<br />
seed extract reduced blood lipid<br />
levels in 38 Chinese human<br />
subjects (Zhejian Zhongyi J. 1995.<br />
30(1):494.” FDA’s letter states<br />
that the preamble to the January 6,<br />
2000 final rule on structure/<br />
function claims gave FDA’s<br />
position on using citations to<br />
scientific references on the product<br />
label or packaging should be<br />
considered a disease claim because<br />
of the unusual and unnecessary<br />
prominence of such placement.<br />
Since the word “cancer” appears in<br />
the citation on the label of the Vigor<br />
Supplier ® Tablet and the claims on<br />
the Radish Seed Tablets includes<br />
disease claims, the articles<br />
expressly or impliedly describe<br />
effects of the ingredients on<br />
diseases. FDA says, since these<br />
claims suggest use as drugs for<br />
treatment, if the claims are used,<br />
the products are subject to<br />
regulation as drugs under the Act.<br />
Docket No. 97S-0163, Ltr 920,<br />
received at the Dockets Office on<br />
March 1, 2007, entered into the<br />
Docket on February (sic) 1, 2007,<br />
and posted to the FDA Website on<br />
March 29, 2007. [NML has marked<br />
the February 1, 2007 dates with<br />
(sic) in the last three summaries<br />
because it seems impossible that the<br />
documents were entered into the files<br />
before they were received. – Ed.]<br />
U.K . RULES -- Continued from page 2...<br />
maintenance of the storage areas<br />
particularly when dust is generated.<br />
“3 Storage of plants, extracts,<br />
tinctures and other preparations may<br />
require special conditions of<br />
humidity, temperature or light<br />
protection; these conditions should be<br />
provided and monitored.<br />
“Production area<br />
“4 Specific provisions should be<br />
taken during sampling, weighing,<br />
mixing and processing operations of<br />
crude plants whenever dust is<br />
generated, to facilitate cleaning and<br />
to avoid cross-contamination, as for<br />
example, dust extraction, dedicated<br />
premises, etc.”<br />
This new publication is the first<br />
thing NML recommends that you<br />
study if you are going to start selling<br />
herbal remedies in the U.K. or<br />
European Community. You can get<br />
more information about this and other<br />
publications of similar nature from<br />
www.medicinescomplete.com where<br />
you will find details about other<br />
publications such as the AHFS Drug<br />
Information publication from the<br />
American Society for Health-System<br />
Pharmacists, the book <strong>Herbal</strong><br />
<strong>Medicine</strong>s, the British National<br />
Formulary, Martindale: The<br />
Complete Drug Reference, and<br />
<strong>Dietary</strong> <strong>Supplement</strong>s that are relied<br />
upon in the U.K. For pricing<br />
information go to:<br />
www.pharmpress.com/shop/<br />
default.asp , the web for the<br />
Pharmaceutical Press.<br />
ONE QUININE PRODUCT<br />
FDA told manufacturers to stop<br />
marketing unapproved drug products<br />
containing quinine because of serious<br />
safety concerns. While quinine has<br />
traditionally been used as a treatment<br />
for malaria, many patients use these<br />
products to treat leg cramps and<br />
similar conditions, a use which is not<br />
approved by FDA.<br />
FDA has received 665 reports of<br />
serious adverse events associated with<br />
quinine use since 1969, including 93<br />
deaths. Among the side effects of<br />
quinine are cardiac arrhythmias,<br />
thrombocytopenia, and severe<br />
hypersensitivity reactions.<br />
Because of the side effects, and<br />
because the toxic dose for quinine is<br />
only slightly higher than the<br />
therapeutic dose, patients should be<br />
warned against treating their leg<br />
cramps with quinine products.<br />
At the present time, Qualaquin,<br />
See QUININE -- Continued on page 18...<br />
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside<br />
Page 18 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
CINCHONA -- Photo courtesy of M L Wee<br />
and The Australian National University.<br />
manufactured<br />
by Mutual<br />
Pharmaceutical<br />
Company, is<br />
the only<br />
quinine product<br />
approved<br />
by the FDA.<br />
Qualaquin is<br />
indicated for<br />
the treatment<br />
of malaria,<br />
and its labeling specifically warns that the product is<br />
not indicated for treating leg cramps. FDA Medwatch<br />
Safety Alert for Quinine products. For more see:<br />
www.fda.gov/medwatch/safety/2006/<br />
safety06.htm#Quinine<br />
CHFA -- continued from page 5...<br />
QUININE -- Cont;d<br />
from page 17...<br />
“Healthy consumers should continue to rely on<br />
antioxidant supplements for the benefits they confer,”<br />
says Wilkie. “This meta analysis does not provide<br />
convincing scientific evidence that antioxidant<br />
supplements do not provide potential health benefits.”<br />
The CHFA reminds consumers that Health Canada<br />
has established monographs with dose information and<br />
indications for the antioxidants beta-carotene, vitamin<br />
A, and vitamin C and has not reported any risks in the<br />
use of these supplements. In fact, the federal government<br />
introduced the <strong>Natural</strong> Health Products <strong>Regulation</strong>s in<br />
2004 to ensure natural health products, such as vitamins,<br />
sold in Canada were safe, had evidence to support their<br />
claims and contained what was on the label.<br />
For further information, contact Natalie Cajic,<br />
Communications Coordinator, at 416-497-6939 ext.<br />
234, 1-800-661-4510 or ncajic@chfa.ca.<br />
The Canadian Health Food Association (CHFA) is<br />
Canada’s largest national trade association<br />
representing the organic and natural products industry.<br />
Its members include over 85 per cent of the subsectors<br />
such as vitamin and mineral supplements,<br />
herbals, homeopathics, sports nutrition products,<br />
natural foods and organic products. CHFA members’<br />
products support Canadians seeking optimal health<br />
and quality of life.<br />
CANADA ADOPTS NEW REGULATIONS<br />
In the monthly update of information concerning<br />
natural health products, the Canadian government has<br />
announced that is has adopted the nutrient content<br />
claims now exiting in the Nutrient Content Claims<br />
listed in sections B.01.500 to B.01.513 and the Table<br />
that follows B.01.513 of the Food and Drug<br />
<strong>Regulation</strong>s. This information can be accessed from<br />
the Department of Justice Canada Website<br />
(www.justice.gc.ca).<br />
The announcement said that after careful<br />
consideration, for the assessment of “absence of” and<br />
“addition of” claims, the NHPD has decided to adopt<br />
these criterion. The Table following B.01.513 in<br />
particular provides definitions of 47 common<br />
“absence of” and “addition of” claims and outlines<br />
specific guidelines and limitations for their use. These<br />
criteria will in turn be used by the NHPD during<br />
product assessments to determine if a specific claim<br />
with respect to the absence or addition of medicinal<br />
or non-medicinal ingredients is true, false or<br />
misleading.<br />
Example:<br />
Based on the Table following section<br />
B.01.513 of the Food and Drug <strong>Regulation</strong>s<br />
For a natural health product to make the claim<br />
“fat-free”, the product must meet the following<br />
criteria:<br />
• less than 0.5 g of fat per reference amount<br />
and serving of stated size; or<br />
• less than 0.5 g of fat per serving of stated<br />
size, if the product is a pre-packaged meal.<br />
If the applicant provides the necessary evidence<br />
demonstrating that the product meets one of the<br />
following criteria listed above, then the following<br />
“absence of” claims would be acceptable: “fat-free”,<br />
“free of fat”, “no fat”, “0 fat”, “zero fat”, “without<br />
fat”, “contains no fat” or “non-fat”<br />
The NHPD will also be developing additional<br />
criteria for “absence of “and “addition of” claims<br />
currently not found in the Table following section<br />
B.01.513. and will deal with these on a case-by-case<br />
basis in the interim.<br />
Also in the new information Health Canada<br />
announced that Revisions have recently been made<br />
See REVISIONS -- Continued on page 19...
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 19<br />
REVISIONS -- Continued from page 18...<br />
to the monographs for Black Cohosh, Ginger, and<br />
Valerian. The revised monographs are currently<br />
available online in the Compendium of Monographs.<br />
From the NHPD Monthly Communiqué, Vol. 2,<br />
Issue 6 - April 2007 found at www.hc-sc.gc.ca/dhpmps/prodnatur/bulletins/communiques/<br />
communique_april07_e.html#1_2<br />
CANADA APPROVES<br />
ASHWAGANDA MONOGRAPH<br />
Health Canada approved a monograph for withania<br />
somniferai on April 18, 2007 for traditional uses in<br />
Ayurveda medicine as a rejuvenative tonic,<br />
convalescence in old age, sleep aid, to balance<br />
aggravated Vata or nervine or tonic, and for memory<br />
enhancement. The dose recommended is 2 to 6 Grams<br />
per day of the dried roots by mouth. The plant is also<br />
known as Ashvagandha, Asgandh and Winter Cherry.<br />
See the full monograph at: www.hc-sc.gc.ca/dhp-mps/<br />
prodnatur/applications/licen-prod/monograph/<br />
mono_ashwagandha_e.html.<br />
The <strong>Natural</strong> Health Products office also modified<br />
the monographs for Blessed Thistle and Heal-All, but<br />
said the changes did not require any action by current<br />
licensees. For details go to: Blessed Thistle at<br />
www.hc-sc.gc.ca/dhp-mps/prodnatur/applications/<br />
licen-prod/monograph/mono_blessedthistlechardonbeni_e.html<br />
and for Heal-All go to: www.hcsc.gc.ca/dhp-mps/prodnatur/applications/licen-prod/<br />
monograph/mono_healall-prunvul_f.html.<br />
AMICUS -- Continued from page 10...<br />
A summary of the argument was presented in the<br />
introduction of the 17 page brief as:<br />
“This case presents an exceptionally important<br />
issue of administrative and statutory law: may<br />
the FDA ignore the clear distinction Congress<br />
has drawn between drugs and dietary<br />
supplements, which are classified as foods,<br />
and thereby greatly expand its authority to<br />
declare a dietary supplement ‘adulterated’<br />
Congress has given the FDA ample powers to<br />
protect public health and safety from<br />
adulterated foods, including dietary<br />
supplements. But in banning ephedrine<br />
alkaloids in dietary supplements, the FDA<br />
chose not to rely on its lawful powers. Instead,<br />
it took the opportunity to re-write the DSHEA,<br />
a statute the FDA strenuously, and<br />
unsuccessfully, opposed.<br />
“The FDA concluded that ephedrine<br />
alkaloids in dietary supplements pose an<br />
‘unreasonable’ risk to health, and are thus<br />
adulterated and should be banned. Amicus<br />
takes no position on whether, under the proper<br />
definition of “unreasonable,” the FDA’s<br />
decision was correct. The FDA relied,<br />
however, on an unauthorized “risk-benefit”<br />
analysis for dietary supplements, essentially<br />
requiring that dietary supplements provide<br />
significant health benefits, proved in rigorous<br />
clinical studies, sufficient to overcome any<br />
risks associated with the product. This is the<br />
standard that governs FDA pre-marketing<br />
approval of drugs; foods, including dietary<br />
supplements, are not subject to any such<br />
standard. The FDA’s test, moreover, affords<br />
the agency an exceptionally large degree of<br />
discretion to ban dietary supplements as<br />
“adulterated.’ Indeed, in applying the standard<br />
in this case, the FDA discounted the<br />
significance of most non-‘health’ benefits<br />
provided by dietary supplements — which are<br />
important to millions of consumers and are<br />
recognized expressly in the DSHEA — and<br />
concluded that those benefits would<br />
henceforth be insufficient to overcome even<br />
a small risk of harm. The District Court held<br />
that this expansive interpretation of the<br />
agency’s powers violated the statute, but the<br />
Tenth Circuit reversed. The FDA’s rule, and<br />
the Tenth Circuit’s decision, cannot be<br />
reconciled with the DSHEA, and a writ of<br />
certiorari is warranted to bring the agency’s<br />
rule in line with Congress’s commands.<br />
See AMICUS -- Continued on page 20...<br />
www.NatMed<strong>Law</strong>.com<br />
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside
Page 20 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside<br />
AMICUS -- Continued from page 19 ...<br />
“Unless this Court grants the petition for a<br />
writ of certiorari, the dietary supplement<br />
industry will suffer severe harm. A significant<br />
percentage of the principal makers of dietary<br />
supplements are located within the Tenth<br />
Circuit, and are therefore subject to its<br />
erroneous decision. If that decision is allowed<br />
to stand, the distinction between dietary<br />
supplements and drugs will effectively<br />
evaporate, the makers of supplements will be<br />
forced to conduct the types of rigorous clinical<br />
tests that Congress declined to require, and<br />
consumer choice in fulfilling their dietary<br />
needs (protected under the DSHEA) will be<br />
curtailed. Amicus urges the Court to grant the<br />
petition.”<br />
The Amicus goes on to say that “The FDA’s<br />
consideration of this issue could, upon application of<br />
a proper standard, result in the banning of EDS”<br />
[ephedra dietary supplements] and broadly covered<br />
the regulatory differences between drugs using a riskbenefit<br />
analysis and foods, particularly DSHEA that<br />
places dietary supplements in the food category. The<br />
“significant or unreasonable” requirement of §<br />
342(f)(1)(A)(i) as interpreted by FDA and the 10 th<br />
Circuit, where more than 150 companies or 20 per<br />
cent of the industry is located in Utah alone, has no<br />
support in the statute or legislative history. The brief<br />
said that “By classifying dietary supplements as foods<br />
…, Congress did not tie the permissibility of selling<br />
supplements to any showing or evidence that they<br />
improve health. Moreover, in permitting the makers<br />
of dietary supplements to make certain types of healthrelated<br />
claims, Congress required only that such<br />
claims be ‘substantiated,’ not clinically proven.”<br />
The Amicus argues that dietary fiber and fish oil,<br />
which are used widely and beneficially, have not been<br />
subjected to the clinical testing that FDA’s new found<br />
risk-benefit standard would require, implying these<br />
would disappear from the market as banned under<br />
the new 10 th Circuit standards. And Amicus told the<br />
Court that the significant problems posed for the<br />
industry by the 10 th Circuit decision will “effectively<br />
unravel the DSHEA.” The Amicus also said that the<br />
10 th Circuit got it wrong when it defined “significant<br />
risk” as a great danger. Under DSHEA “substantial”<br />
and “unreasonable” define two different categories<br />
of harm in the context of dietary supplements. Amicus<br />
challenged the Court to allow it to help “prove, on<br />
the merits, that the FDA has overreached and<br />
exceeded the bounds of Congress’ statutory scheme.”<br />
Joseph R. Guerra, Esq. is Counsel of Record, with<br />
I. Scott Bass, Esq. and Robert A. Parker, Esq. of Sidley<br />
Austin LLP of Washington, D.C. (202) 736-8000 on<br />
this Amicus Curiae brief.<br />
HERBAL PRODUCT MAKERS<br />
SAY DROP THE GUIDANCE<br />
In the March 2007 issue of <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong><br />
Newsletter the FDA’s Guidance on Complementary<br />
and Alternative <strong>Medicine</strong> Products and Their<br />
<strong>Regulation</strong> by the Food and Drug Administration was<br />
summarized, but not explored. The FDA first<br />
proposed the Guidance on February 27. Now the<br />
American <strong>Herbal</strong> Products Association (AHPA) of<br />
Silver Spring, Maryland has filed comments telling<br />
FDA to drop the whole idea. It looks like the FDA<br />
Office of Policy and Planning came up with an<br />
unnecessary document. AHPA wants the document<br />
to go away.<br />
In fairly pointed and direct remarks, the comments<br />
of AHPA on the Guidance said that the group “has<br />
observed that, since publication of the draft guidance,<br />
confusion has mounted and continues to grow. This<br />
confusion extends to the content of the draft, as well<br />
as to FDA’s purpose in developing the draft. The result<br />
is that publication of the draft guidance has had<br />
exactly the opposite effect as the agency’s expressed<br />
intention to address confusion concerning the<br />
regulatory status of products used by practitioners of<br />
complementary and alternative medicine. AHPA<br />
strongly encourages the agency to discontinue any<br />
further substantive work on this topic. Rather, FDA<br />
should simply inform the public that the draft<br />
guidance has been withdrawn.”<br />
This was predictable to NML since most people<br />
selling products that interface with complementary<br />
and alternative medicine have never studied food and<br />
drug law. Also some health care professionals have<br />
become enraged at the FDA’s attempt to control and<br />
See CAM GUIDANCE -- Continued on page 21...
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 21<br />
CAM GUIDANCE -- Continued from page 20...<br />
define the practice of their roles, taking regulation<br />
from the states to the FDA. On this issue AHPA said<br />
“AHPA notes that FDA does not regulate the practice<br />
of medicine and does not have jurisdiction over health<br />
care practitioners, whether conventional, allopathic,<br />
complementary or alternative.”<br />
Also FDA failed to state in the Guidance who the<br />
document was meant to control, said AHPA. When<br />
FDA cited reasons for issuing the Guidance, it stated<br />
two: 1.) That FDA “has seen increased confusion as<br />
to whether certain products used in CAM… are<br />
subject to regulation under the Federal Food, Drug,<br />
and Cosmetic Act (‘the Act’) or Public Health Service<br />
Act (‘PHS Act’).” 2.) That the agency has “also seen<br />
an increase in the number of CAM products imported<br />
into the United States.”<br />
But AHPA said it might be that FDA employees are<br />
confused and instead of a Guidance to Industry, there<br />
may be a need to issue a new training manual to FDA<br />
employees. And AHPA added, “if it were<br />
representatives of the media that are confused, for<br />
example, by identifying products used by CAM<br />
practitioners as “unregulated,” FDA could and should<br />
issue specific clarifying statements, but again should<br />
not rely on a guidance for industry to address media<br />
confusion; and that if it is the public that is confused,<br />
FDA could, for example, address this confusion with<br />
one or more articles in FDA Consumer magazine, but<br />
not through guidance for industry.”<br />
AHPA also said that FDA has ignored Executive<br />
Order 13422. On January 18, 2007 President Bush<br />
signed Executive Order 13422, amending Executive<br />
Order 12866 on Regulatory Planning and Review of<br />
September 30, 1993. Of significance to these<br />
comments is that the amendments enacted on January<br />
18, 2007 expanded the scope of this Executive Order<br />
to place restrictions on federal regulatory agencies<br />
not only when they promulgate regulations but also<br />
when they issue guidance documents. Specifically,<br />
the draft guidance document raises ‘novel legal or<br />
policy issues arising out of legal mandates,’ this<br />
triggering requirements under the Executive Order.<br />
These requirements have not been met.”<br />
Finally AHPA said that the announcement was<br />
inconsistent with its dates for comments, saying in<br />
one place that comments must be in within 90 days<br />
of publication and in another stating that the date was<br />
April 30, 2007, a date that was only 62 days after<br />
publication. If 90 days is the operative date, then<br />
May 28, 2007 is the date comments are due. [It<br />
appears that someone at FDA was permitted to issue<br />
the Guidance without knowing how to submit it to<br />
the Federal Register. – Ed.] The full AHPA<br />
comments are available at: www.ahpa.org/Portals/0/<br />
pdfs/07_0427_AHPAComments_FDA_CAM<br />
_Guidance.pdf/.<br />
As of May 14, 2007, the FDA had posted less than<br />
10 of the 55 comments to the public website, including<br />
the AHPA comments, which means that the only<br />
people who know what is going on with this issue are<br />
those who have personal researchers in the FDA<br />
Docket Clerks Office on a daily basis to extract and<br />
copy records. Comment 55 was placed on the FDA<br />
web and updated April 20, 2007.<br />
TRADEMARKS FOR<br />
ÆSCULUS HIPPOCASTANUM<br />
Surprise ! There are no trademarks on the USPTO<br />
web based data base that contain the words<br />
“ÆSCULUS HIPPOCASTANUM,” but as readers of<br />
NLM know well every Latin binomial herb has a<br />
common name. Neither are the words “Common<br />
Horse Chestnut” found in the USPTO trademark files.<br />
When carefully thought about, then searched, one can<br />
find three “dead” marks for “Horse Chestnut,” and<br />
no marks at all for “Common Horse” or “Common<br />
Chestnut.”<br />
Therefore, it may be possible to obtain a trademark<br />
for “Horse Chestnut.” But for what The last three<br />
were for dietary supplements, but they were either<br />
cancelled or abandoned, most likely because the<br />
products did not sell and did not warrant the expense<br />
of registration.<br />
Horse Chestnut Plus was filed March 9, 1999 as a<br />
mark for dietary supplements by Metagenics, Inc. of<br />
San Clemente, California in International Class 005.<br />
The mark was first used January 28, 1999. But the<br />
See TRADEMARKS -- Continued on page 22...<br />
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside
Copyright 2007 by Muscatatuck Publishers, Inc. For Information on photocopying, see Page 2 Inside<br />
Page 22 May 2007 <strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM<br />
TRADEMARKS -- Cont’d from p.21...<br />
application was cancelled under<br />
Section 8 for failure to file an<br />
affidavit of use on January 6, 2007.<br />
See Serial No. 75/660962.<br />
Horse Chestnut Xtra was filed<br />
March 16, 1998 as a mark for<br />
vitamins, minerals, dietary and<br />
nutritional supplements containing<br />
horse chestnut by Rexall Sundown,<br />
Inc. of Boca Raton, Florida in<br />
International Class 005. This mark<br />
was filed as an intent to use mark<br />
and was apparently never used,<br />
since the mark was abandoned<br />
November 19, 1999. See Serial<br />
No. 75/451206.<br />
Horse Chestnut-Power was<br />
filed by Twin Laboratories, Inc. of<br />
American Fork, Utah, on July 29,<br />
1997 for dietary supplements, and<br />
herbs, extracts, concentrates, and<br />
combinations thereof, all for use as<br />
dietary supplements. Twin Labs<br />
assigned the mark to ISI Brands,<br />
Inc. of Grand Rapids, Michigan,<br />
but the mark was cancelled<br />
February 25, 2006 under Section<br />
8, which means no affidavit of use<br />
was filed in the time required. See<br />
Serial No. 75/332373.<br />
What was so magical about this<br />
name in 1997 to 1999 And why<br />
did all three companies drop the<br />
products. NML does not know.<br />
But read the article about Horse<br />
Chestnut in the Wellness Letter and<br />
what NML has added to it. You will<br />
learn that Horse Chestnut is used<br />
widely in Europe for varicose veins<br />
and hemorrhoids. Now remember<br />
your DSHEA. Rubbing Horse<br />
Chestnut on your legs or around<br />
your bottom is not “ingestion” and<br />
therefore these products could not<br />
be dietary supplements under<br />
DSHEA.<br />
Still interested in using this Horse<br />
Chestnut on a product Then do a<br />
“Google” search and one of the<br />
first files that comes up is one<br />
listing thirteen studies that<br />
appeared in the medical literature<br />
from 1979 to 1999 from an article<br />
from Environmental Nutrition<br />
newsletter by Denise Webb, Ph.D.<br />
See www.horsechestnut.com for<br />
the list of articles. Maybe you will<br />
want to get this trademark for an<br />
OTC drug. This might take some<br />
more research.<br />
Another page you can find on the<br />
web is a page from http://<br />
nccam.nih.gov/health/<br />
horsechestnut/ that is on the<br />
National Institutes of Health<br />
website. Here you will again see<br />
recommendations for the use of<br />
products made from Horse<br />
Chestnut for venious insufficiency<br />
and hemorrhoids, but it is not<br />
recommended for anything else.<br />
There are a number of Horse<br />
Chestnut seed extracts on the<br />
market, some standardized to<br />
contain certain amounts of Aescin<br />
or esculin, the ingredient thought<br />
to be the active ingredient.<br />
Keep in mind that words used for<br />
trademarks may be “descriptive”<br />
under the statute and not<br />
immediately registerable. But over<br />
time a descriptive name can<br />
become known in the market place<br />
as the source for the product, in<br />
which case it can become<br />
distinctive and registerable. Talk<br />
to your legal counsel about better<br />
ways to make a trademark<br />
distinctive.<br />
WWW.NATMEDLAW.COM<br />
CONE FLOWER (E. Intermedia)<br />
Photo Courtesy of Lloyd Library and Museum<br />
DS GMPs -- Continued from page 1...<br />
estimate) expected the release of<br />
the GMPs in December 2006.<br />
If you are still worried about what<br />
to do, go to 68 Federal Register<br />
12157 to review the last Notice of<br />
Proposed Rule Making and read<br />
what FDA wants you to do. Then<br />
you will understand why OMB is<br />
taking so long.<br />
NEW INGREDIENTS -- Cont’d from p. 9...<br />
to the FDA Website on November<br />
20, 2006 by kk.<br />
National Center for Tobacco<br />
Research of Sarasota, Florida, sent<br />
FDA an NDI, dated June 21, 2006<br />
for a natural herbal extract,<br />
cytisine, from the Golden Rain<br />
plant (Cytsius labroinu L.) grown<br />
in Italy and other European countries.<br />
The supplier is a Bulgarian<br />
company, Sopharma, that sells a<br />
product named Tabex. Each tablet<br />
contains 1.5 mg of cytisine and it<br />
is used for various indications including<br />
smoking cessation. The<br />
See NEW INGREDIENTS -- Cont’d on p. 23... .
<strong>Natural</strong> <strong>Medicine</strong> <strong>Law</strong> TM May 2007 Page 23<br />
NEW INGREDIENTS -- Conti’d from page 22...<br />
usual dose is one tablet four times<br />
a day. With the letter was an abstract<br />
of a clinical study of 436<br />
consecutive attendees of a smoking<br />
clinic in Warsaw, Poland, that<br />
tested Tabex and the conclusions<br />
were that the results were similar<br />
to that of nicotine replacement<br />
therapy. The article was apparently<br />
not published as no citation was<br />
given. FDA responded by letter<br />
of September 7, 2006 to Kirk G.<br />
Voelker, M.D., from Linda S.<br />
Pellicore, Ph.D., stating that the<br />
notification does not comply with<br />
21 CFR 190.6 and is incomplete<br />
as it did not contain an original and<br />
two copies, it did not provide a<br />
description of the dietary supplement<br />
that contains the new ingredient,<br />
and a history of use or other<br />
evidence of safety was not provided.<br />
FDA said it was unable to<br />
determine on the basis of the one<br />
clinical study that was provided<br />
whether it provides an adequate<br />
basis for a conclusion that the dietary<br />
supplement will reasonably<br />
be expected to be safe because the<br />
information is incomplete. FDA<br />
said if the product is marketed<br />
without an adequate notification it<br />
will be considered adulterated and<br />
introduction of such a product in<br />
interstate commerce is prohibited.<br />
Docket No. 1995S-0316, RPT 357,<br />
received at the Dockets Office on<br />
October 6, 2006, filed October 6,<br />
2006, and posted to the FDA<br />
Website on November 20, 2006 by<br />
kk.<br />
RPT 358 is not on the Public<br />
Website.<br />
SCIENTIFIC WELLNESS of<br />
DOX, LLC of Ann Arbor, Michigan,<br />
filed a NDI, dated June 26, 2007,<br />
for 19-nordehydro-epiandrosterone<br />
made in 100 mg capsules to be<br />
taken three times a day with the<br />
directions “DIRECTIONS FOR<br />
USE: This product is for adults<br />
over the age of 21 only. Do not<br />
exceed recommended dosage.<br />
This product is not intended to diagnose,<br />
treat, cure or prevent any<br />
disease.” KEEP OUT OF REACH<br />
OF CHILDREN. The information<br />
given indicates that DHEA was<br />
marketed before October 15, 1994.<br />
The letter was not signed but had<br />
attached numerous pages discussing<br />
DHEA. FDA responded by<br />
letter of September 19, 2007, from<br />
Linda S. Pellicore, Ph.D. to Dennis<br />
Hayes begins by acknowledging<br />
DHEA, but at the last paragraph<br />
on page one, states that<br />
“Your notification about ‘Nucleic<br />
Acid <strong>Supplement</strong> Capsules’ does<br />
not comply with the requirements<br />
of 21 CFR 190.6 and is incomplete.”<br />
FDA points out the letter<br />
was not signed and only abstracts<br />
were submitted. FDA indicated it<br />
had requested three copies of all<br />
reference articles and did not receive<br />
them. Because of this, FDA<br />
is unable to proceed with the review.<br />
FDA’s response never made<br />
clear why it started the response<br />
with one NDI and completed it<br />
with another as the subject NDI.<br />
Docket No. 1995S-0316, RPT 359,<br />
received at the Dockets Office on<br />
November 3, 2006, filed November<br />
3 and 12, 2006, and posted to<br />
the FDA Website on February 12,<br />
2007.<br />
AJINOMOTO U.S.A., Inc. of<br />
Washington, D.C. sent a NDI to<br />
FDA, dated July 18, 2006, concerning<br />
a product named<br />
Capsinoids (Extracted Oil of Sweet<br />
Chili Peppers) also called CH-19<br />
Sweet Extract. It is derived from<br />
Capsicum annuum L. chili peppers.<br />
Soft gelatin capsules containing 1<br />
mg will be recommended to take<br />
three 1 mg capsules orally once per<br />
day. [FDA did not post the<br />
company’s Notice. - Ed.] FDA responded<br />
to this notice by letter of<br />
October 2, 2006 stating that it had<br />
“significant concerns” about the<br />
evidence presented. FDA was unable<br />
to determine the identity of the<br />
ingredient because the extraction<br />
process if given only in very general<br />
terms. The notice does not include<br />
methods of analysis, active<br />
or inactive. The notice does not<br />
provide studies with the preparation<br />
that will be marketed. It is<br />
unclear, FDA said, how Capsiate<br />
Natura is similar to peppers and<br />
this unclear how information about<br />
its use is related to the NDI. The<br />
history of use of chili peppers<br />
throughout the world given in the<br />
notice, FDA said, gives estimated<br />
current intakes of capsinoids in the<br />
U.S. per capita. “However, the recommended<br />
intake of the new dietary<br />
ingredient (i.e., 3 mg<br />
capsinoids/day) is about twice the<br />
current daily intake at the 90 th percentile<br />
of intake by chili pepper<br />
eaters. Moreover, it is not possible<br />
to calculate the concentration of<br />
capsinoids present in the proposed<br />
dietary supplement based upon the<br />
information provided.” FDA mentioned<br />
several other reasons and<br />
concluded that the information<br />
does not provide an adequate basis<br />
to conclude that the product will<br />
reasonable be expected to be safe<br />
when used as suggested. FDA said<br />
that introduction of the product into<br />
interstate commerce is prohibited.<br />
Docket No. 1995S-0316, RPT 360,<br />
received at the Dockets Office on<br />
November 3, 2006, filed November<br />
3 and 12, 2006, and posted to the<br />
FDA Website on February 12, 2007.<br />
Copyright, 2007 by Muscatatuck Publishers, Inc. For Information on Photocopying, see Page 2 Inside
PHARMACY LAW<br />
SEMINAR<br />
Developments in Pharmacy <strong>Law</strong><br />
Seminar XVIII will be November<br />
8 - 11, 2007 at Loews Coronado<br />
Bay Resort, Coronado, California.<br />
Details are available at the American<br />
Society for Pharmacy <strong>Law</strong><br />
website -- www.aspl.org. The program<br />
is cosponsored by the National<br />
Association of Chain Drug<br />
Stores.<br />
MAY IS NATIONAL HIGH<br />
BLOOD PRESSURE<br />
EDUCATION MONTH<br />
The National Center for Complementary<br />
and Alternatiave <strong>Medicine</strong><br />
is promoting the month on<br />
http://nccam.nih.gov<br />
Check out the recommendations<br />
about natural medicines. One in 3<br />
Americans has high blood pressure.<br />
The 15th Annual Symposium featuring<br />
20 speakers will be held June<br />
1 to 4 at the Blue Ridge Assembly<br />
in Black Mountain, North Carolina.<br />
Registration information can be<br />
obtained at (800) 252-0688 and<br />
and www.botanicalmedicine.org.<br />
AMERICAN HOLISTIC<br />
MEDICAL ASSOCIATION<br />
AHMA 2007 Clinical & Scientific<br />
Conference, Portland, Oregon,<br />
June 6 - 9, 2007. Details are<br />
www.holisticmedicine.org or The<br />
Center for Mind-Body <strong>Medicine</strong>,<br />
5225 Connecticut Ave, NW, #414,<br />
Washington, D.C. 20015. All<br />
health professions are invited.<br />
National Pharmacy <strong>Law</strong> and<br />
Ethics Symposium<br />
Sponsored by the American Society<br />
for Pharmacy <strong>Law</strong> and the<br />
Duquesne University School of<br />
Pharmacy. June 8-9, 2007<br />
Pittsburgh, PA. Rooms are reserved<br />
at the Marriott Pittsburgh<br />
City Center hotel, one block from<br />
Hanley Hall where the symposium<br />
will be held. For more info go to<br />
www.aspl.org.<br />
HARVESTING HEALTH<br />
Denial of Certiorari in the ephedra<br />
case will increase the use of the<br />
legal ephedra tea for about the next<br />
10 years until another case is decided<br />
differently from that in the<br />
10th Circuit. Then the “de novo”<br />
review provisions in the law that<br />
were not applied by the10th Circuit<br />
might be read the way the Congress<br />
wanted. Or the Congress might<br />
strengthen DSHEA by amending<br />
the review provisions. NML will<br />
watch this issue carefully.<br />
The U. S. Senate passed a bill<br />
giving FDA more responsibility.<br />
See the Special <strong>Supplement</strong> in this<br />
issue. If the House agrees, will either<br />
body properly fund the new<br />
tasks, or will FDA have to struggle<br />
to figure out its priorities<br />
The new Reagan-Udall Institute<br />
will be faulted in a few years, particularly<br />
after a director is appointed,<br />
and that person testifies a<br />
few times before dozens of committees<br />
of Congress. There will<br />
little time for this new “chief scientist”<br />
to coordinate research activities.<br />
The current FDA Commissioner<br />
will become more like a Chief of<br />
Police in the public mind. That is<br />
what the Editor believes could happen<br />
down the road.<br />
The European Community and<br />
other states are fine tuning their<br />
laws concerning dietary supplements<br />
and herbal medicines.<br />
Developments on most of the<br />
World’s seven continents will continue<br />
in the dietary supplement and<br />
related fields.<br />
This issue marks the end of 10<br />
years for this<br />
newsletter.<br />
Thanks to all<br />
who keep<br />
reading.<br />
need their help.<br />
Let others<br />
know about<br />
us because<br />
you will soon<br />
William J. Skinner, R.Ph.,<br />
Attorney at <strong>Law</strong>, Editor




