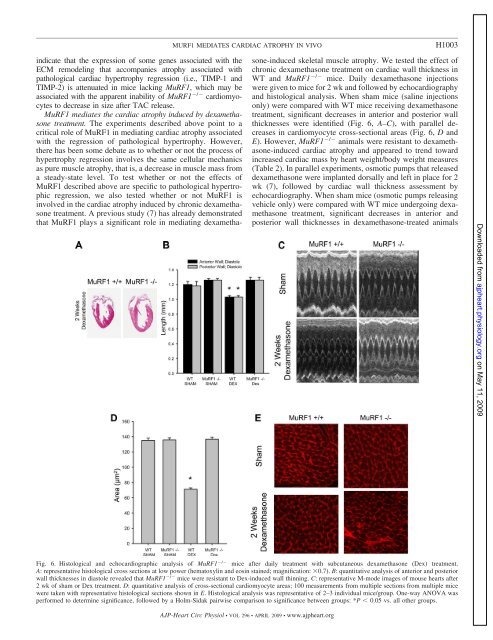
MURF1 MEDIATES CARDIAC ATROPHY IN VIVOH1003indicate that the expression of some genes associated with theECM remodeling that accompanies atrophy associated withpathological cardiac hypertrophy regression (i.e., TIMP-1 <strong>and</strong>TIMP-2) is attenuated in mice lacking MuRF1, which may beassociated with the apparent inability of MuRF1 / cardiomyocytesto decrease in size after TAC release.MuRF1 mediates the cardiac atrophy induced by dexamethasonetreatment. The experiments described above point to acritical role of MuRF1 in mediating cardiac atrophy associatedwith the regression of pathological hypertrophy. However,there has been some debate as to whether or not the process ofhypertrophy regression involves the same cellular mechanicsas pure muscle atrophy, that is, a decrease in muscle mass froma steady-state level. To test whether or not the effects ofMuRF1 described above are specific to pathological hypertrophicregression, we also tested whether or not MuRF1 isinvolved in the cardiac atrophy induced by chronic dexamethasonetreatment. A previous study (7) has already demonstratedthat MuRF1 plays a significant role in mediating dexamethasone-inducedskeletal muscle atrophy. We tested the effect ofchronic dexamethasone treatment on cardiac wall thickness inWT <strong>and</strong> MuRF1 / mice. Daily dexamethasone injectionswere given to mice for 2 wk <strong>and</strong> followed by echocardiography<strong>and</strong> histological analysis. When sham mice (saline injectionsonly) were compared with WT mice receiving dexamethasonetreatment, significant decreases in anterior <strong>and</strong> posterior wallthicknesses were identified (Fig. 6, A–C), with parallel decreasesin cardiomyocyte cross-sectional areas (Fig. 6, D <strong>and</strong>E). However, MuRF1 / animals were resistant to dexamethasone-inducedcardiac atrophy <strong>and</strong> appeared to trend towardincreased cardiac mass by heart weight/body weight measures(Table 2). In parallel experiments, osmotic pumps that releaseddexamethasone were implanted dorsally <strong>and</strong> left in place for 2wk (7), followed by cardiac wall thickness assessment byechocardiography. When sham mice (osmotic pumps releasingvehicle only) were compared with WT mice undergoing dexamethasonetreatment, significant decreases in anterior <strong>and</strong>posterior wall thicknesses in dexamethasone-treated animalsDownloaded from ajpheart.physiology.org on May 11, 2009Fig. 6. Histological <strong>and</strong> echocardiographic analysis of MuRF1 / mice after daily treatment with subcutaneous dexamethasone (Dex) treatment.A: representative histological cross sections at low power (hematoxylin <strong>and</strong> eosin stained; magnification: 0.7). B: quantitative analysis of anterior <strong>and</strong> posteriorwall thicknesses in diastole revealed that MuRF1 / mice were resistant to Dex-induced wall thinning. C: representative M-mode images of mouse hearts after2 wk of sham or Dex treatment. D: quantitative analysis of cross-sectional cardiomyocyte areas; 100 measurements from multiple sections from multiple micewere taken with representative histological sections shown in E. Histological analysis was representative of 2–3 individual mice/group. One-way ANOVA wasperformed to determine significance, followed by a Holm-Sidak pairwise comparison to significance between groups: *P 0.05 vs. all other groups.AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org
H1004MURF1 MEDIATES CARDIAC ATROPHY IN VIVOTable 2. Transthoracic echocardiography on unanesthetized MuRF1 / <strong>and</strong> WT control mice after 2 wk of dailysubcutaneous saline or dexamethasone treatmentSaline TreatmentDexamethasone TreatmentWT MuRF1 / WT MuRF1 /n 9 9 8 6Heart rate, beats/min 581.322.5 614.912.1 585.815.7 596.813.1Anterior wall at diastole, mm 1.200.02 1.260.02 1.030.02* 1.260.04Posterior wall at diastole, mm 1.180.04 1.190.01 0.970.01* 1.180.01Anterior wall at systole, mm 1.830.04 1.960.05 1.430.03 1.860.02Posterior wall at systole, mm 1.700.04 1.580.03 1.440.03* 1.670.03LVEDD 2.80.4 3.40.3 3.00.1 3.20.1LVESD 1.30.2 1.50.2 1.40.1 1.50.1LV mass/body weight, mg/g 3.990.19 4.040.19 3.320.12* 5.470.18*LV mass/tibia length, mg/mm 6.880.34 7.430.40 5.870.22* 9.060.41*Heart weight/body weight, mg/g 5.200.28 5.740.25 4.180.07* 6.010.25Fractional shortening, % 54.51.0 55.00.9 52.81.8 54.80.8Ejection fraction, % 86.50.90 86.50.7 84.71.5 86.40.6Values are means SE; n, no. of mice/group. LVEDD, left ventricular (LV) end-diastolic dimension; LVESD, LV end-systolic dimension. LV mass wascalculated as follows: (external LV diameter in diastole 3 LVEDD 3 ) 1.055. Fractional shortening was calculated as follows: (LVEDD LVESD)/LVEDD 100. The ejection fraction was calculated as follows: (end Simpson’s diastolic volume end Simpson’s systolic volume)/end Simpson’s diastolic volume 100.*P 0.05 vs. all other groups.were identified, as anticipated (Supplemental Fig. 4), alongwith cardiac dilation (Supplemental Table 2). In contrast,MuRF1 / mice were resistant to dexamethasone-induced cardiacatrophy, as indicated by little or no changes in anterior <strong>and</strong>posterior wall thicknesses in diastole (Supplemental Fig. 4) orchamber dilation (Supplemental Table 2). Although it wasnoted that the osmotic pump experiments demonstrated agreater overall atrophy than the daily dexamethasone injections,technical issues with wound dehiscence (likely due to thedexamethasone treatment) made further analysis of this observationimpractical. These findings suggest that MuRF1 mediatesatrophy in the dexamethasome model in the same mannerthat it does in the atrophy associated with pathological cardiachypertrophy regression. MuRF1 may therefore play a moregeneralized role in decreasing cardiac muscle mass in a varietyof clinical scenarios.DISCUSSIONThe development of pathological cardiac hypertrophy is acommon precursor to heart failure <strong>and</strong> heightens the risk ofheart failure <strong>and</strong> arrhythmias. Not surprisingly, the reversal ofpathological cardiac hypertrophy reduces these risks <strong>and</strong> istherefore an attractive process against which to target potentialtherapies. In the present study, we used two models of cardiacatrophy to investigate the role of MuRF1 in decreasing cardiacmuscle mass in vivo. The concept of cardiac atrophy in thepresent study has been exp<strong>and</strong>ed from muscle mass loss frombaseline levels to the decrease in muscle mass seen in thetherapeutic regression of hypertrophic states. Our results demonstratethat MuRF1 is an essential mediator of the cardiacatrophy associated with both regression of TAC-inducedpathological hypertrophy as well as the atrophy resulting fromchronic dexamethasone treatment. Together, these findingsdemonstrate, for the first time, a major role for MuRF1 in theprocess of reducing cardiac muscle mass in vivo. As preclinicaltrials have demonstrated the value of blunting hypertrophicgrowth without compromising cardiac performance, the potentialfor antihypertrophy therapy has been suggested (19, 20).This strongly supports the notion of exploring MuRF1 as auseful therapeutic target in the quest to improve clinical outcomes<strong>and</strong> prevent heart failure in a wide range of patients.While the reversal of pathological cardiac hypertrophy thatoccurs after the removal of pressure overload appears toinvolve a reduction in cardiomyocyte size, it is only one aspectof a broader process that involves the restoration of diastolicfunction <strong>and</strong> remodeling of the ECM. In clinical scenarioswhere high blood pressure is adequately treated or aorticstenosis is surgically repaired, reversal of pathological cardiachypertrophy occurs in patients. In this situation, several studieshave reported that regression of pathological cardiac hypertrophyparallels improvements in diastolic dysfunction (12, 22,30, 46). A balance of degradation by MMPs <strong>and</strong> TIMPs in theECM is vital to the remodeling process that occurs duringhypertrophy regression. In failing hearts, alterations in thebalance of MMPs <strong>and</strong> their endogenous inhibitors (TIMPs)have been reported. The regulation of MMPs <strong>and</strong> TIMPsduring the regression of pathological cardiac hypertrophy,which occurs in as little as 4 days in the present study, has notbeen previously reported. We identified that WT mice had anexpected transient increase in MMP-2, TIMP-1, TIMP-2, <strong>and</strong>ColI mRNA levels 4 days after TAC release. In contrast, thesedid not change in MuRF1 / mice. While we don’t necessarilybelieve that MuRF1 has a direct effect on ECM regulation,MuRF1’s effect on cardiomyocyte size might allow it toindirectly affect the ECM.In the present study, we identified, for the first time, thatdexamethasone treatment in adult mice leads to a reduction incardiac mass. This should be contrasted to the effects that dexamethasonehas on the heart in both human <strong>and</strong> experimentalneonates. Dexamethasone therapy in neonates for bronchpulmonarydysplasia, premature birth, <strong>and</strong> chronic lung disease has beenreported to be associated with increased cardiac mass repeatedly(23, 38, 41, 48, 52). In a r<strong>and</strong>omized clinical trial, a common sideeffect of dexamethasone therapy is cardiac hypertrophy aftertherapy for as little as 7 days, resulting in clinically significantsymptoms (54). Therefore, the effects of dexamethasone-inducedcardiac atrophy reported in the present study are observationslikely confined to adult mouse hearts. The role of MuRF1 in thisDownloaded from ajpheart.physiology.org on May 11, 2009AJP-Heart Circ Physiol • VOL 296 • APRIL 2009 • www.ajpheart.org