

JAK2-V617F mutation in a patient with Philadelphia - Mannheimer ...
JAK2-V617F mutation in a patient with Philadelphia - Mannheimer ...
JAK2-V617F mutation in a patient with Philadelphia - Mannheimer ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
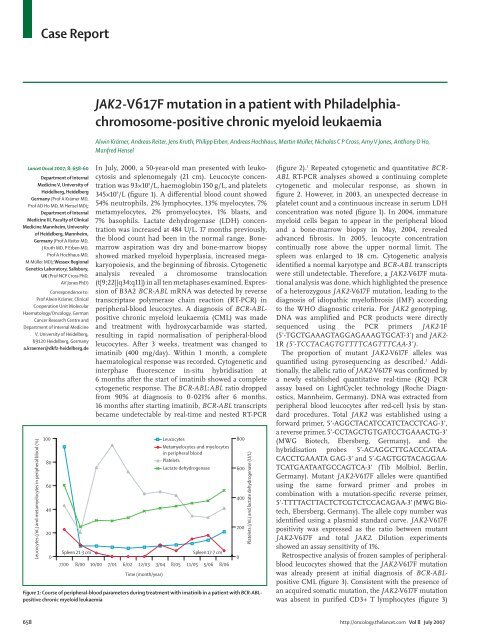
Case Report<strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> <strong>in</strong> a <strong>patient</strong> <strong>with</strong> <strong>Philadelphia</strong>chromosome-positivechronic myeloid leukaemiaAlw<strong>in</strong> Krämer, Andreas Reiter, Jens Kruth, Philipp Erben, Andreas Hochhaus, Mart<strong>in</strong> Müller, Nicholas C P Cross, Amy V Jones, Anthony D Ho,Manfred HenselLancet Oncol 2007; 8: 658–60Department of InternalMedic<strong>in</strong>e V, University ofHeidelberg, HeidelbergGermany (Prof A Krämer MD,Prof AD Ho MD, M Hensel MD);Department of InternalMedic<strong>in</strong>e III, Faculty of Cl<strong>in</strong>icalMedic<strong>in</strong>e Mannheim, Universityof Heidelberg, Mannheim,Germany (Prof A Reiter MD,J Kruth MD, P Erben MD,Prof A Hochhaus MD,M Müller MD); Wessex RegionalGenetics Laboratory, Salisbury,UK (Prof NCP Cross PhD,AV Jones PhD)Correspondence to:Prof Alw<strong>in</strong> Krämer, Cl<strong>in</strong>icalCooperation Unit MolecularHaematology/Oncology, GermanCancer Research Centre andDepartment of Internal Medic<strong>in</strong>eV, University of Heidelberg,69120 Heidelberg, Germanya.kraemer@dkfz-heidelberg.deLeucocytes (/nL) and metamyelocytes <strong>in</strong> peripheral blood (%)100806040200Spleen 21·3 cmIn July, 2000, a 50-year-old man presented <strong>with</strong> leukocytosisand splenomegaly (21 cm). Leucocyte concentrationwas 93×10 9 /L, haemoglob<strong>in</strong> 150 g/L, and platelets345×10 9 /L (figure 1). A differential blood count showed54% neutrophils, 2% lymphocytes, 13% myelocytes, 7%metamyelocytes, 2% promyelocytes, 1% blasts, and7% basophils. Lactate dehydrogenase (LDH) concentrationwas <strong>in</strong>creased at 484 U/L. 17 months previously,the blood count had been <strong>in</strong> the normal range. Bonemarrowaspiration was dry and bone-marrow biopsyshowed marked myeloid hyperplasia, <strong>in</strong>creased megakaryopoiesis,and the beg<strong>in</strong>n<strong>in</strong>g of fibrosis. Cytogeneticanalysis revealed a chromosome translocation(t[9;22][q34;q11]) <strong>in</strong> all ten metaphases exam<strong>in</strong>ed. Expressionof B3A2 BCR-ABL mRNA was detected by reversetranscriptase polymerase cha<strong>in</strong> reaction (RT-PCR) <strong>in</strong>peripheral-blood leucocytes. A diagnosis of BCR-ABLpositivechronic myeloid leukaemia (CML) was madeand treatment <strong>with</strong> hydroxycarbamide was started,result<strong>in</strong>g <strong>in</strong> rapid normalisation of peripheral-bloodleucocytes. After 3 weeks, treatment was changed toimat<strong>in</strong>ib (400 mg/day). With<strong>in</strong> 1 month, a completehaem atological response was recorded. Cytogenetic and<strong>in</strong>terphase fluorescence <strong>in</strong>-situ hybridisation at6 months after the start of imat<strong>in</strong>ib showed a completecytogenetic response. The BCR-ABL:ABL ratio droppedfrom 90% at diagnosis to 0·021% after 6 months.16 months after start<strong>in</strong>g imat<strong>in</strong>ib, BCR-ABL transcriptsbecame undetectable by real-time and nested RT-PCRLeucocytesMetamyelocytes and myelocytes<strong>in</strong> peripheral bloodPlateletsLactate dehydrogenaseSpleen 17·7 cm7/00 8/00 10/00 7/01 6/02 12/03 3/04 8/05 11/05 5/06 8/06Time (month/year)Figure 1: Course of peripheral-blood parameters dur<strong>in</strong>g treatment <strong>with</strong> imat<strong>in</strong>ib <strong>in</strong> a <strong>patient</strong> <strong>with</strong> BCR-ABLpositivechronic myeloid leukaemia8006004002000Platelets (/nL) and lactate dehydrogenase (U/L)(figure 2). 1 Repeated cytogenetic and quantitative BCR-ABL RT-PCR analyses showed a cont<strong>in</strong>u<strong>in</strong>g completecytogenetic and molecular response, as shown <strong>in</strong>figure 2. However, <strong>in</strong> 2003, an unexpected decrease <strong>in</strong>platelet count and a cont<strong>in</strong>uous <strong>in</strong>crease <strong>in</strong> serum LDHconcentration was noted (figure 1). In 2004, immaturemyeloid cells began to appear <strong>in</strong> the peripheral bloodand a bone-marrow biopsy <strong>in</strong> May, 2004, revealedadvanced fibrosis. In 2005, leucocyte concentrationcont<strong>in</strong>ually rose above the upper normal limit. Thespleen was enlarged to 18 cm. Cytogenetic analysisidentified a normal karyotype and BCR-ABL transcriptswere still undetectable. Therefore, a <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong>alanalysis was done, which highlighted the presenceof a heterozygous <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong>, lead<strong>in</strong>g to thediagnosis of idiopathic myelofibrosis (IMF) accord<strong>in</strong>gto the WHO diagnostic criteria. For <strong>JAK2</strong> genotyp<strong>in</strong>g,DNA was amplified and PCR products were directlysequenced us<strong>in</strong>g the PCR primers <strong>JAK2</strong>-1F(5’-TGCTGAAAGTAGGAGAAAGTGCAT-3’) and <strong>JAK2</strong>-1R (5’-TCCTACAGTGTTTTCAGTTTCAA-3’).The proportion of mutant <strong>JAK2</strong>-<strong>V617F</strong> alleles wasquantified us<strong>in</strong>g pyrosequenc<strong>in</strong>g as described. 2 Additionally,the allelic ratio of <strong>JAK2</strong>-<strong>V617F</strong> was confirmed bya newly established quantitative real-time (RQ) PCRassay based on LightCycler technology (Roche Diagnostics,Mannheim, Germany). DNA was extracted fromperipheral blood leucocytes after red-cell lysis by standardprocedures. Total <strong>JAK2</strong> was established us<strong>in</strong>g aforward primer, 5’-AGGCTACATCCATCTACCTCAG-3’,a reverse primer, 5’-CCTAGCTGTGATCCTGAAACTG-3’(MWG Biotech, Ebersberg, Germany), and thehybridisation probes 5’-ACAGGCTTGA CCCATAA-CACCT GAAATA GAG-3’ and 5’-GAGTGGTACAGGAA-TCATGAATAATGCCAGTCA-3’ (Tib Molbiol, Berl<strong>in</strong>,Germany). Mutant <strong>JAK2</strong>-<strong>V617F</strong> alleles were quantifiedus<strong>in</strong>g the same forward primer and probes <strong>in</strong>comb<strong>in</strong>ation <strong>with</strong> a <strong>mutation</strong>-specific reverse primer,5’-TTTTACTTACTCTCGTCTCCACAGAA-3’ (MWG Bio -tech, Ebersberg, Germany). The allele copy number wasidentified us<strong>in</strong>g a plasmid standard curve. <strong>JAK2</strong>-<strong>V617F</strong>positivity was expressed as the ratio between mutant<strong>JAK2</strong>-<strong>V617F</strong> and total <strong>JAK2</strong>. Dilution experimentsshowed an assay sensitivity of 1%.Retrospective analysis of frozen samples of peripheralbloodleucocytes showed that the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong>was already present at <strong>in</strong>itial diagnosis of BCR-ABLpositiveCML (figure 3). Consistent <strong>with</strong> the presence ofan acquired somatic <strong>mutation</strong>, the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong>was absent <strong>in</strong> purified CD3+ T lymphocytes (figure 3)658 http://oncology.thelancet.com Vol 8 July 2007
Case Reportand buccal cells (data not shown). As demonstrated bytwo <strong>in</strong>dependent methods—<strong>JAK2</strong>-<strong>V617F</strong> pyrosequenc<strong>in</strong>gand PCR—the proportion of the mutant allele stayedroughly constant from diagnosis of CML <strong>in</strong> July, 2000, toFebruary, 2006 (figure 2). Repeated cytogenetic analysis<strong>in</strong> May, 2006 aga<strong>in</strong> showed a normal karyotype.<strong>JAK2</strong> is a tyros<strong>in</strong>e k<strong>in</strong>ase that has an important role <strong>in</strong>the signall<strong>in</strong>g pathways of many haemopoietic growthfactorreceptors. The s<strong>in</strong>gle acquired po<strong>in</strong>t <strong>mutation</strong><strong>V617F</strong> (1849G>T) <strong>in</strong> <strong>JAK2</strong> occurs <strong>in</strong> 50–97% of <strong>patient</strong>s<strong>with</strong> IMF, essential thrombocytosis, and polycythemiavera. 2–6 By contrast, the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> has neverbeen identified <strong>in</strong> a <strong>patient</strong> <strong>with</strong> BCR-ABL-positiveCML. 7 This is the first description of a <strong>patient</strong>, to ourknowledge, <strong>in</strong> whom the BCR-ABL fusion gene and anacquired somatic <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> could bedetected contemporaneously, <strong>with</strong> IMF becom<strong>in</strong>gcl<strong>in</strong>ically relevant after successful treatment of CML byimat<strong>in</strong>ib.Several conclusions can be drawn from these data.First, bone-marrow fibrosis was not a consequence ofprogressive CML. Rather, myelofibrosis developed as asecond myeloproliferative disorder (MPD) dur<strong>in</strong>gcomplete cytogenetic and molecular remission of CML.Although the BCR-ABL fusion gene might represent the<strong>in</strong>itiat<strong>in</strong>g lesion <strong>in</strong> most cases of CML, cl<strong>in</strong>icalmanifestations of the disease can be variable. Specifically,bone-marrow fibrosis can occur, sometimes vary<strong>in</strong>ggreatly dur<strong>in</strong>g the course of the disease, and has beenused to categorise <strong>patient</strong>s <strong>in</strong>to different groups <strong>with</strong>dist<strong>in</strong>ctive survival caracter istics. 8 Fibrogenesis <strong>in</strong> MPDis generally assumed to be mediated by the abnormalrelease of transform<strong>in</strong>g growth factor-β and plateletderivedgrowth factor (PDGF). Imat<strong>in</strong>ib, a selective<strong>in</strong>hibitor of ABL, KIT, and PDGF receptor tyros<strong>in</strong>ek<strong>in</strong>ases, and which is not active aga<strong>in</strong>st <strong>JAK2</strong>, has beenseen to reduce the content of bone-marrow fibre <strong>in</strong><strong>patient</strong>s <strong>with</strong> CML. 9 Regression of myelofibrosis byimat<strong>in</strong>ib is thought to be caused either by a direct PDGFantagonisticeffect or by normalisation of megakaryopoiesisfrom which abnormal concentrations of PDGFare released. How ever, expression of <strong>JAK2</strong>-<strong>V617F</strong> <strong>in</strong>mur<strong>in</strong>e haematopoietic cells leads to MPD associated<strong>with</strong> myelofibrosis. 10 Moreover, expression of a BCR-<strong>JAK2</strong>fusion gene has been described <strong>in</strong> a <strong>patient</strong> <strong>with</strong> atypicalCML and bone-marrow fibrosis, 11 and the PCM1-<strong>JAK2</strong>fusion is associated <strong>with</strong> the development of myelofibrosis<strong>in</strong> <strong>patient</strong>s <strong>with</strong> chronic and acute leukaemias. 12 Thesef<strong>in</strong>d<strong>in</strong>gs suggest that <strong>JAK2</strong>-<strong>V617F</strong> contributes to thedevelopment of myelofibrosis <strong>in</strong> MPD, 3,10 and might havebeen responsible for the <strong>in</strong>duction of myelofibrosis <strong>in</strong>our <strong>patient</strong> which, consequently, was <strong>in</strong>dependent of thepresence of BCR-ABL and did not respond to imat<strong>in</strong>ibtreatment. This assumption is supported by the absenceof cl<strong>in</strong>ical and molecular responses of <strong>patient</strong>s <strong>with</strong> IMFand polycythemia vera treated <strong>with</strong> imat<strong>in</strong>ib. 13,14 Iftreatment <strong>with</strong> imat<strong>in</strong>ib had not been <strong>in</strong>itiated, thecl<strong>in</strong>ical course of the <strong>patient</strong> could only have beenspeculated upon. Treatment and response to imat<strong>in</strong>ibmight have accelerated the outgrowth of the IMF.Second, our data suggest that <strong>in</strong> our <strong>patient</strong>, the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> had occurred before the acquisition ofthe <strong>Philadelphia</strong> (Ph)-chromosome. Development of aPh-positive CML has rarely been reported <strong>in</strong> <strong>patient</strong>s<strong>with</strong> MPD. 15 In the few cases that have been reported,ACPercentage of tumour cells100958075605540352015Figure 2: BCR-ABL:ABL ratio and <strong>JAK2</strong>-<strong>V617F</strong> allele frequency dur<strong>in</strong>g treatment <strong>with</strong> imat<strong>in</strong>ib <strong>in</strong> a <strong>patient</strong><strong>with</strong> BCR-ABL-positive chronic myeloid leukaemiaParameters established by pyrosequenc<strong>in</strong>g and PCR.A0T G TA T G TTGTGT C T GT C T GTime (month/year)BDA T G T G T C T GA T G TBCR-ABL:ABL<strong>JAK2</strong>/<strong>V617F</strong>-pyrosequenc<strong>in</strong>g<strong>JAK2</strong>/<strong>V617F</strong>-PCR7/00 7/01 7/02 7/03 7/04 7/05 7/06TGT C T GFigure 3: Presence of the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> <strong>in</strong> a <strong>patient</strong> <strong>with</strong> BCR-ABL-positive chronic myeloid leukaemiaA heterozygous G→T transversion <strong>in</strong> <strong>JAK2</strong> (arrows) is present <strong>in</strong> DNA from leucocytes (A,C,D) at different timepo<strong>in</strong>ts (A: July, 2000; C: November, 2001; D: May, 2006) dur<strong>in</strong>g the course of the disease, but absent <strong>in</strong> T cells(B; May, 2006), consistent <strong>with</strong> the existence of an acquired somatic orig<strong>in</strong> of the <strong>mutation</strong>.http://oncology.thelancet.com Vol 8 July 2007 659
Case Reportthere is a debate as to whether MPD and CML ariseseparately from each other, represent<strong>in</strong>g <strong>in</strong>dependenttransformation of a normal stem cell, or whether the Phchromosomearises <strong>in</strong> a stem cell that was part of theMPD clone. In our <strong>patient</strong>, <strong>JAK2</strong>-<strong>V617F</strong> was detectable atan almost constant allele frequency <strong>in</strong> all peripheralbloodsamples exam<strong>in</strong>ed (figure 2). The most likelyexplanation for this f<strong>in</strong>d<strong>in</strong>g is the presence of aheterozygous <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> <strong>in</strong> most of theperipheral-blood leucocytes, implicat<strong>in</strong>g the simultaneouspresence of the BCR-ABL fusion gene and the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> <strong>in</strong> white blood cells. Because BCR-ABLbecame undetectable after treatment <strong>with</strong> imat<strong>in</strong>ib, butthe <strong>JAK2</strong>-<strong>V617F</strong> allele frequency rema<strong>in</strong>ed virtuallyunchanged, the conclusion can be drawn that the Phchromosomewas acquired from a haematopoietic cellcarry<strong>in</strong>g the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong>. Several f<strong>in</strong>d<strong>in</strong>gssuggest that MPD are caused by a <strong>mutation</strong> <strong>in</strong> an, as yet,unknown gene that precedes the acquisition of either a<strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> or a BCR-ABL fusion gene. 3,16 Themost reasonable explanation for the f<strong>in</strong>d<strong>in</strong>gs presentedhere is that BCR-ABL can, <strong>in</strong> rare cases, occur on thebackground of the <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong>, possiblyaugmented by an elusive <strong>in</strong>itial <strong>mutation</strong> predispos<strong>in</strong>g tothe acquisition of both a <strong>JAK2</strong>-<strong>V617F</strong> <strong>mutation</strong> and aBCR-ABL translocation.Conflicts of <strong>in</strong>terestThe authors declared no conflicts of <strong>in</strong>terest.AcknowledgmentsWe thank Brigitte Schreiter for excellent technical assistance. This workwas supported by a grant from the German José Carreras LeukemiaFoundation (DJCLS R 06/04).References1 Emig M, Saussele S, Wittor H, et al. Accurate and rapid analysis ofresidual disease <strong>in</strong> <strong>patient</strong>s <strong>with</strong> CML us<strong>in</strong>g specific fluorescenthybridization probes for real time quantitative RT-PCR. Leukemia1999; 13: 1825–32.2 Jones AV, Kreil S, Zoi K, et al. Widespread occurrence of the <strong>JAK2</strong><strong>V617F</strong> <strong>mutation</strong> <strong>in</strong> chronic myeloproliferative disorders. Blood 2005;106: 2162–68.3 Kralovics R, Passamonti F, Buser AS, et al. A ga<strong>in</strong>-of-function<strong>mutation</strong> of <strong>JAK2</strong> <strong>in</strong> myeloproliferative disorders. N Engl J Med2005; 352: 1779–90.4 Baxter EJ, Scott LM, Campbell PJ, et al. Acquired <strong>mutation</strong> of thetyros<strong>in</strong>e k<strong>in</strong>ase <strong>JAK2</strong> <strong>in</strong> human myeloproliferative disorders.Lancet 2005; 365: 1054–61.5 James C, Ugo V, Le Couedic JP, et al. A unique clonal <strong>JAK2</strong><strong>mutation</strong> lead<strong>in</strong>g to constitutive signall<strong>in</strong>g causes polycythaemiavera. Nature 2005; 434: 1144–48.6 Lev<strong>in</strong>e RL, Wadleigh M, Cools J, et al. Activat<strong>in</strong>g <strong>mutation</strong> <strong>in</strong> thetyros<strong>in</strong>e k<strong>in</strong>ase <strong>JAK2</strong> <strong>in</strong> polycythemia vera, essentialthrombocythemia, and myeloid metaplasia <strong>with</strong> myelofibrosis.Cancer Cell 2005; 7: 387–97.7 Jel<strong>in</strong>ek J, Oki Y, Gharibyan V, et al. <strong>JAK2</strong> <strong>mutation</strong> 1849G>T is rare<strong>in</strong> acute leukemias but can be found <strong>in</strong> CMML, <strong>Philadelphia</strong>chromosome-negative CML, and megakaryocytic leukemia.Blood 2005; 106: 3370–73.8 Kvasnicka HM, Thiele J, Schmitt-Graeff A, et al. Bone marrowfeatures improve prognostic efficiency <strong>in</strong> multivariate riskclassification of chronic-phase Ph(1+) chronic myelogenousleukemia: a multicenter trial. J Cl<strong>in</strong> Oncol 2001; 19: 2994–3009.9 Beham-Schmid C, Apfelbeck U, Sill H, et al. Treatment of chronicmyelogenous leukemia <strong>with</strong> the tyros<strong>in</strong>e k<strong>in</strong>ase <strong>in</strong>hibitor STI571results <strong>in</strong> marked regression of bone marrow fibrosis. Blood 2002;99: 381–83.10 Lacout C, Pisani DF, Tulliez M, Moreau Gachel<strong>in</strong> F, Va<strong>in</strong>chenker W,Villeval JL. <strong>JAK2</strong><strong>V617F</strong> expression <strong>in</strong> mur<strong>in</strong>e hematopoietic cellsleads to MPD mimick<strong>in</strong>g human PV <strong>with</strong> secondary myelofibrosis.Blood 2006; 108: 1652–60.11 Gries<strong>in</strong>ger F, Hennig H, Hillmer F, et al. A BCR-<strong>JAK2</strong> fusion geneas the result of a t(9;22)(p24;q11.2) translocation <strong>in</strong> a <strong>patient</strong> <strong>with</strong> acl<strong>in</strong>ically typical chronic myeloid leukemia. Genes ChromosomesCancer 2005; 44: 329–33.12 Reiter A, Walz C, Watmore A, et al. The t(8;9)(p22;p24) is arecurrent abnormality <strong>in</strong> chronic and acute leukemia that fusesPCM1 to <strong>JAK2</strong>. Cancer Res 2005; 65: 2662–67.13 Tefferi A, Mesa RA, Gray LA, et al. Phase 2 trial of imat<strong>in</strong>ibmesylate <strong>in</strong> myelofibrosis <strong>with</strong> myeloid metaplasia. Blood 2002;99: 3854–56.14 Jones AV, Silver RT, Waghorn K, et al. M<strong>in</strong>imal molecular response<strong>in</strong> polycythemia vera <strong>patient</strong>s treated <strong>with</strong> imat<strong>in</strong>ib or <strong>in</strong>terferonalpha. Blood 2006; 107: 3339–41.15 Curt<strong>in</strong> NJ, Campbell PJ, Green AR. The <strong>Philadelphia</strong> translocationand pre-exist<strong>in</strong>g myeloproliferative disorders. Br J Haematol 2005;128: 734–36.16 Kralovics R, Teo SS, Li S, et al. Acquisition of the <strong>V617F</strong> <strong>mutation</strong>of <strong>JAK2</strong> is a late genetic event <strong>in</strong> a subset of <strong>patient</strong>s <strong>with</strong>myeloproliferative disorders. Blood 2006; 108: 1377–80.660 http://oncology.thelancet.com Vol 8 July 2007




