TECNICHE DI BIOLOGIA MOLECOLARE - Il Saturatore
TECNICHE DI BIOLOGIA MOLECOLARE - Il Saturatore
TECNICHE DI BIOLOGIA MOLECOLARE - Il Saturatore

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
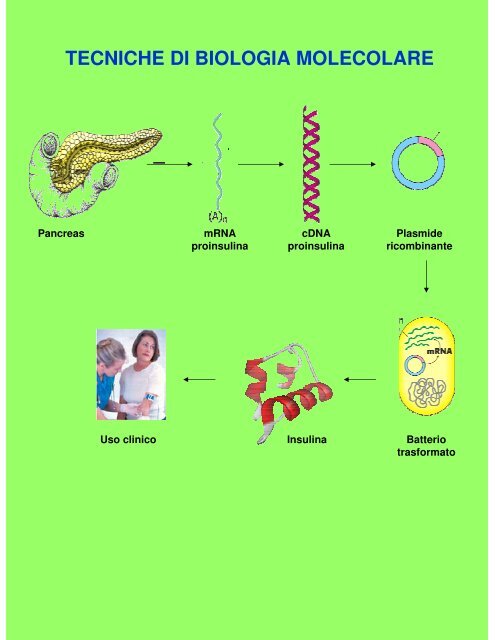
<strong>TECNICHE</strong> <strong>DI</strong> <strong>BIOLOGIA</strong> <strong>MOLECOLARE</strong><br />
Pancreas mRNA<br />
proinsulina<br />
Uso clinico<br />
cDNA<br />
proinsulina<br />
Plasmide<br />
ricombinante<br />
Insulina Batterio<br />
trasformato
<strong>Il</strong> DNA ricombinante: gli enzimi di restrizione<br />
Si dicono nucleasi gli enzimi che idrolizzano un legame<br />
fosfodiesterico di una molecola di DNA o RNA, generalmente<br />
in corrispondenza di una sequenza specifica di nucleotidi.<br />
Le nucleasi vengono distinte in nucleasi per RNA e quelle per<br />
DNA.<br />
Nell’ambito di quelle per il DNA, distinguiamo poi le<br />
esonucleasi, che possono tagliare solo il nucleotide ad<br />
un’estremità della molecola (ad esempio una 3’-5’<br />
endonucleasi del veleno di serpenti), dalle endonucleasi, che<br />
operano invece tagli all’interno di una doppia elica di DNA.
Distinguiamo tre tipi di endonucleasi per DNA:<br />
• tipo I: riconoscono un sito e operano un taglio lontano da esso, in<br />
una posizione variabile.<br />
• tipo II: riconoscono un sito e operano un taglio al suo interno, in<br />
una posizione specifica.<br />
tipo III: presentano un diverso meccanismo di azione, ma hanno in<br />
comune con le endonucleasi di tipo I la mancanza di specificità di<br />
taglio.<br />
La rottura in siti specifici della doppia elica di DNA è un sistema di<br />
difesa di molti procarioti nei confronti di DNA estraneo che riesca<br />
ad entrare nella cellula (ad esempio, il DNA di un fago). In questo<br />
modo si contrasta l’infezione della cellula e se ne preserva<br />
l’integrità del genoma.
<strong>Il</strong> sistema delle nucleasi non è specifico per un invasore<br />
piuttosto che per un altro: si basa sulla probabilità che un lungo<br />
tratto di DNA contenga la sequenza riconosciuta dal pool di<br />
nucleasi della cellula. Tuttavia, il DNA di un procariota contiene<br />
a sua volta migliaia di basi ed è possibile che il sito di<br />
restrizione riconosciuto dalle sue nucleasi sia presente anche<br />
nel suo genoma.<br />
Com’è possibile che venga tagliato il DNA esogeno e non<br />
quello endogeno?<br />
<strong>Il</strong> sistema di difesa basato sulle endonucleasi è complementare<br />
a un sistema di riconoscimento del DNA endogeno basato sulla<br />
metilazione: in un dato organismo, è presente, per ogni enzima<br />
di restrizione, una metiltransferasi in grado di metilare un<br />
nucleotide sullo stesso sito riconosciuto dall’endonucleasi. I siti<br />
così metilati sono protetti dall’azione delle endonucleasi.
Sono invece tagliati i tratti di DNA estraneo appena entrano nella<br />
cellula.<br />
Se un fago riesce comunque ad entrare e infettare la cellula<br />
ospite, il genoma virale subisce lo stesso pattern di metilazione<br />
del DNA cellulare, risultanto così refrattario alla digestione da<br />
parte delle endonucleasi.<br />
Se si duplica ed esce dalla cellula potrà facilmente infettare<br />
batteri dello stesso tipo, dei quali può contrastare il sistema di<br />
difesa, ma non organismi o ceppi che posseggano un sistema di<br />
endonucleasi/metiltransferasi diverso.<br />
Si dice quindi che il fago è ristretto ad un determinato ceppo, da<br />
cui il nome “enzimi di restrizione” dato alle endonucleasi.<br />
Nelle endonucleasi di tipo I e III la metiltransferasi è un dominio<br />
della stessa proteina, mentre per quelle di tipo II l’attività è svolta<br />
da una proteina diversa
In virtù della loro specificità di taglio, le endonucleasi di tipo II<br />
sono quelle di gran lunga più interessanti per le applicazioni<br />
biotecnologiche.<br />
La maggior delle endonucleasi di tipo II riconosce sequenze<br />
specifiche di 4, 5 o 6 nucleotidi. Caratteristica comune a questi<br />
siti è il fatto di essere quasi tutti palindromici (o “a simmetria<br />
binaria”).
La simmetria binaria del sito di riconoscimento è<br />
giustificata dalla simmetria binaria dell’enzima stesso,<br />
generalmente in forma dimerica: data la struttura della<br />
doppia elica, nucleotidi su filamenti opposti distanziati di<br />
circa 5 unità si trovano sullo stesso lato della doppia elica.
Vi sono due possibili tagli: quelli che lasciano estremità blunt<br />
(taglio “pari” sui due filamenti) e quelli che lasciano estremità<br />
sticky (con un numero di basi diverso sui due filamenti). I<br />
nucleaotidi “sporgenti” prendono il nome di “overhangs”.<br />
Ciascun enzima di restizione è caratterizzato:<br />
1. Dal sito di restrizione riconosciuto<br />
2. Dalla modalità di taglio (blunt, stiky)
Data la grande importanza biotecnologica delle endonucleasi<br />
di tipo II, si è cercato di isolarne quante più possibile. Oggi ne<br />
sono note migliaia, di cui diverse centinaia sono in<br />
commercio. <strong>Il</strong> nome, per convenzione, deriva dall’organismo<br />
dal quale è stato isolato.<br />
Gli enzimi di restrizione che riconoscono un identico sito,<br />
anche se non lo tagliano nella stessa posizione, sono detti<br />
isoschizomeri.
DNA ricombinante<br />
L’impiego in vitro di due classi di enzimi – gli enzimi di<br />
restrizione e la ligasi – ha consentito di sviluppare la<br />
tecnologia del DNA ricombinante. Due frammenti di DNA<br />
duplex tagliati con il medesimo enzima di restrizione che dia<br />
un taglio “sticky” avranno overhangs complementari che, in<br />
opportune condizioni, tenderanno ad appaiarsi.
La ligasi, a spese dell’ATP, potrà formare due legami<br />
fosfodiesterici. Una volta formato il legame covalente, il<br />
DNA ricombinante è stabile. Si noti che si mantiene il sito di<br />
restrizione, per cui i due frammenti uniti potranno sempre<br />
essere separati per digestione con l’enzima di restrizione<br />
corrispondente (in questo caso BamHI).<br />
Se una delle due molecole tagliate è circolare (ad esempio<br />
un plasmide), la reazione di “taglia e cuci” per<br />
restrizione/ligazione porta, in due passaggi, un DNA<br />
circolare.
La ligasi catalizza anche la formazione di due legami covalenti<br />
tra due estremità blunt, tra le quali, ovviamente, non vi ci può<br />
essere riconoscimento tra overhangs. Come prevedibile, la<br />
reazione richiede condizione più drastiche e tempi più lunghi.<br />
A meno che non sia stato usato la stessa endonucleasi per<br />
rendere blunt i due frammenti, il sito di restrizione viene a<br />
perdersi.<br />
La ligazione di estremità blunt, per quanto meno efficiente, è<br />
più generale, in quanto le estremità blunt possono essere<br />
generate con enzimi di restrizione diversi, o addirittura<br />
“smussando”, con enzimi specifici (polishing enzymes), le<br />
estremità sticky. <strong>Il</strong> prodotto PCR di alcune DNA polimerasi è<br />
blunt. Inoltre, grazie al fatto che il legame indipendentemente<br />
dalla sequenza dei frammenti coinvolti, si possono legare corti<br />
tratti di DNA duplex sintetici a qualsiasi estremità blunt di<br />
frammenti ottenuti in altro modo.
Separazione di polinucleotidi<br />
Per poter analizzare il DNA, è cruciale<br />
disporre di metodi che consentano la<br />
separazione di frammenti di diversa<br />
lunghezza. Tutti i metodi di separazione<br />
del DNA si basano sul principio<br />
dell’elettroforesi: si applica un campo<br />
elettrico alla miscela da risolvere, posta<br />
in una matrice che fa da “setaccio<br />
molecolare”.<br />
<strong>Il</strong> DNA, a pH neutro, presenta caica<br />
netta negativa, per cui tutti i frammenti<br />
tenderanno a migrare verso il catodo.<br />
La velocità di migrazione è funzione<br />
della lunghezza del frammento. Alla fine<br />
della corsa i frammenti di diversa<br />
lunghezza si disporranno quindi in<br />
bande discrete.<br />
Per frammenti lunghi fino a 1000 bp si<br />
impiega il gel di poliacrilamide (PAGE),<br />
mentre per frammenti più grandi si usa<br />
il più poroso gel di agarosio. Applicando<br />
campi elettrici pulsati ai gel di agarosio<br />
si possono separare interi cromosomi.<br />
<strong>Il</strong> DNA non subisce danni durante la<br />
migrazione, per cui lo si può facilmente<br />
recuperare in forma pura tagliando la<br />
banda corrispondente al frammento che<br />
interessa (elettroforesi preparativa).
Le bande di DNA possono essere visualizzate in vari modi. <strong>Il</strong><br />
metodo aspecifico più usato consiste nel trattare il gel con<br />
bromuro di etidio, una sostanza che diventa fluorescente solo<br />
quando lega il DNA. <strong>Il</strong>luminando il gel con una radiazione di<br />
opportuna lunghezza d’onda, si osserverà quindi luce di<br />
fluorescenza in corrispondenza delle bande.<br />
Si può far correre la miscela che si vuole risolvere parallelamente<br />
a una miscela di markers, ovvero a frammenti di DNA di<br />
lunghezza nota. Ciò consente di stimare la lunghezza delle<br />
diverse bande.
<strong>Il</strong> blotting<br />
Proprio perché porosi, i gel di agarosio non sono adatti a<br />
mantenere a lungo le bande discrete di DNA: i frammenti, in<br />
breve tempo, tendono a diffondere. In seguito alla<br />
separazione su gel, è però possibile trasferire le bande di<br />
DNA su un supporto di nitrocellulosa, sul quale viene<br />
preservata la distribuzione dei frammenti ottenuta per<br />
elettroforesi anche per tempi lunghi.<br />
Si ottiene quindi una replica stabile del gel, che consente<br />
l’applicazione di diverse tecniche di riconoscimento dei<br />
frammenti.
<strong>Il</strong> Southern blotting<br />
<strong>Il</strong> Southern blotting (dal nome dello scopritore), consente di<br />
identificare il frammento di DNA nel quale si trovi la sequenza<br />
complementare a una sonda sintetica marcata<br />
radioattivamente.<br />
Ad esempio, un intero genoma può essere digerito con<br />
enzimi di restrizione e fatto migrare in gel di agarosio. Un<br />
singolo frammento può essere individuato mediante southern<br />
blotting.<br />
<strong>Il</strong> blotting viene condotto in ambiente fortemente basico, in<br />
modo che il DNA si denaturi e si separino i due filamenti che<br />
lo costituiscono.<br />
I filamenti così separati possono così ibridizzare (formazione<br />
di un breve tratto di doppi elica) con una sequenza di DNA o<br />
RNA (sintetica) marcata radioattivamente che sia<br />
complementare. Un’autoradiografia consente di individuare la<br />
banda che contiene il tratto complementare alla nostra<br />
sonda.<br />
5’ – GGGCCCTTTAAAATCGCA ATGTTAACGGGGCCCTTTAAAATC........TGTTTAAACCCGGGTTT TAA TTTAAACCCGGGTTT-3’<br />
3’ – CCCCGGGAAATTTTAGCGATACAATTGCCCCGGGAAATTTTAG.......AC.AAATTTGGGCCCAAAATT AAATTTGGGCCCAAA 5’<br />
5’ – GGGCCCTTTAAAATCGCA ATGTTAACGGGGCCCTTTAAAATC........TGTTTAAACCCGGGTTTTAATTTAAACCCGGGTTT -3’<br />
3’ - TACAATTGCCCCGGGAAATTT<br />
3’ - TACAATTGCCCCGGGAAATTT
La rilevazione di un frammento di DNA e le sue dimensioni<br />
possono essere importanti per diversi scopi, ad esempio<br />
la diagnosi di una malattia genetica.
PCR<br />
La reazione di polimerizzazione a catena (polymerase<br />
chain reaction, PCR) è una tecnica di amplificazione in<br />
vitro di un tratto specifico di DNA.<br />
<strong>Il</strong> materiale di partenza può essere una quantità minima di<br />
DNA, generalmente il materiale genetico estratto da una<br />
cultura cellulare, da un campione di tessuto, ecc.
In vivo:<br />
L’enzima che catalizza l’aggiunta di un nucleotide alla catena<br />
in formazione è la DNA polimerasi.<br />
Esistono diverse DNA polimerasi, ma sono tutte accomunate<br />
da una forma a “mano” grazie alla quale la proteina può<br />
“avvolgere” il DNA.
La polimerizzazione dei nucleotidi avviene a partire dai loro<br />
derivati trifosfato. La rottura del legame fosfoanidridico<br />
fornisce l’energia per formare il legame fosfoesterico con<br />
l’ossidrile in posizione 3’ del nucleotide successivo.<br />
<strong>Il</strong> deossinucleotide aggiunto è, tra i 4 che costituiscono il<br />
DNA, quello complementare al filamento stampo.
Le DNA polimerasi non possono iniziare la sintesi del<br />
filamento complementare ex novo: non possono cioè partire<br />
da un singolo filamento, possono solo allungare una doppia<br />
elica, anche molto corta.<br />
Interviene pertanto un enzima che riesce a partire da un<br />
singolo filamento e sintetizzare un corto tratto di RNA (non<br />
DNA) che formi una doppia elica con il filamento singolo.<br />
Questo corto “innesco” per la DNA polimerasi è detto primer.<br />
L’enzima primasi è una RNA polimerasi specializzata che si<br />
unisce al complesso del pre-innesco e sintetizza un primer di<br />
RNA complementare al frammento da duplicare.<br />
Perché un frammento di RNA e non uno direttamente di DNA?<br />
perché le RNA polimerasi, contrariamente alle DNA<br />
polimerasi, possono partire da un filamento singolo e non<br />
richiedono un primer. Evoluzionisticamente, non si è cioè<br />
sviluppata una DNA polimerasi che possa partire da un singolo<br />
filamento e si è quindi affermato questo sistema più<br />
complesso, che richiede un ulteriore passaggio. <strong>Il</strong> corto tratto a<br />
RNA sarà infatti eliminato in seguito da enzimi specifici.
I primers di RNA saranno poi rimossi e rimpiazzati dai<br />
corrispondenti filamenti in DNA da una DNA polimerasi.<br />
Segmenti adiaceti saranno poi legati covalentemente<br />
dall’enzima ligasi
La DNA polimerasi richiede che la doppia elica di DNA<br />
venga localmente aperta da parte di proteine specifiche.<br />
Inizialmente, l’apertura della doppia elica interessa poche<br />
centinaia di nucleotidi, che formano la bolla di replicazione. In<br />
questa regione i filamenti singoli sono esposti e può essere<br />
iniziata la duplicazione.<br />
L’apertura della doppia elica a livello della forcella di<br />
replicazione viene catalizzata da enzimi noti come elicasi.<br />
I tratti a filamento singolo che si generano vengono protetti<br />
da specifiche proteine (single strand DNA binding proteins o<br />
SSB)
La forcella di replicazione in E. coli è quindi un sistema<br />
complesso che vede la partecipazione di un elevato numero<br />
di proteine
Nella PCR:<br />
La PCR sfrutta la capacità della DNA polimerasi di<br />
sintetizzare la catena complementare a un filamento<br />
singolo di DNA in presenza dei 4 deossinucleotidi<br />
trifosfato e di adeguati primer.<br />
Rispetto al processo di trascrizione che avviene in vivo,<br />
dove il distacco tra i due filamenti della doppia elica è<br />
mediato da enzimi, nella PCR si sfrutta il fenomeno della<br />
denaturazione termica. La doppia elica del DNA tende a<br />
dissociarsi nei due filamenti costituenti ad una temperatura di<br />
circa 95°C.<br />
Essendo richiesta una fase ad alta temperatura, non è<br />
possibile impiegare le DNA polimerasi degli organismi più<br />
comuni, inattivate dal calore. La soluzione è utilizzare una<br />
DNA-polimerasi termostabile, isolata da organismi che<br />
normalmente vivono ad alte temperature (ad esempio<br />
Thermus aquaticus, da cui Taq polimerasi, la prima ad<br />
essere stata usata). La Taq polimerasi è attiva attorno a<br />
72°C e resistente alla denaturazione fino ad oltre i 95°C.
La Taq polimerasi, come tutte le DNA polimerasi, necessita<br />
di un tratto a doppia elica sul quale si possa innestare l’elica<br />
complementare allo stampo.<br />
Affinchè la polimerasi agisca, occorre quindi un primer (uno<br />
per filamento), un corto segmento di DNA a singolo filamento<br />
che si appai al DNA che si vuole amplificare formando un<br />
breve tratto a doppia elica.<br />
I primer, che ovviamente devono essere di sequenza nota e<br />
complementare alla catena con cui li si vuole appaiare, sono<br />
ottenuti per sintesi. Le tecnologie attuali consentono di<br />
sintetizzare tratti di DNA di lunghezza fino a 100 nucleotidi<br />
(molto più corti di un gene: per questo non si possono<br />
sintetizzare direttamente i geni, anziché applicare la PCR).<br />
Le sequenze corrispondenti ai primers sono l’unica parte del<br />
DNA da amplificare che deve essere nota in partenza.<br />
5’ – GGGCCCTTTAAAATCGCA ATGTTAACGGGGCCCTTTAAAATC........TGTTTAAACCCGGGTTT TAA TTTAAACCCGGGTTT-3’<br />
3’ – CCCCGGGAAATTTTAGCGATACAATTGCCCCGGGAAATTTTAG.......AC.AAATTTGGGCCCAAAATT AAATTTGGGCCCAAA 5’<br />
5’ – GGGCCCTTTAAAATCGCA ATGTTAACGGGGCCCTTTAAAATC........TGTTTAAACCCGGGTTTTAATTTAAACCCGGGTTT -3’<br />
3’ - TACAATTGCCCCGGGAAATTT<br />
TTTAAACCCGGGTTTTAA -3’<br />
3’- CCCCGGGAAATTTTAGCGATACAATTGCCCCGGGAAATTTTAG........ ACAAATTTGGGCCCAAAATTTTTAAACCCGGGTTT - 5’
Nel complesso, nella miscela di reazione (generalmente<br />
pochi µl di soluzione) occorreranno quindi:<br />
• DNA da amplificare. <strong>Il</strong> tratto che interessa può in realtà<br />
essere la minima parte di quello presente, spesso l’intero<br />
genoma.<br />
• La Taq polimerasi o uno dei diversi enzimi analoghi che<br />
sono stati isolati e messi in commercio e i suoi cofattori (ioni<br />
Mg 2+ ).<br />
• I quattro deossinucleotidi trifosfato, che costituiscono il<br />
substrato della DNA polimerasi anche in vivo.<br />
• i 2 primer che si appaino agli estremi delle due catene<br />
complementari nel tratto che si vuole amplificare. I primer<br />
devono essere aggiunti in grande eccesso, affinchè siano<br />
sufficienti per tutti i cicli.
La miscela di reazione può quindi essere incubata a 95°C per<br />
denaturare il DNA (30 secondi o più). La si porta poi a circa 55°C<br />
per favorire l’appaiamento delle catene singole con i primer e<br />
infine a circa 72°C, la temperatura alla quale la Taq polimerasi è<br />
più attiva. Gli steps di temperatura sono generati da uno<br />
strumento detto thermocycler.<br />
I tempi e le temperature per ognuno dei passaggi dipende in<br />
realtà da una quantità di fattori: il tipo di polimerasi usata, la<br />
lunghezza del segmento da amplificare, la natura dei primers, ecc.<br />
Un ciclo tipo è il seguente:<br />
circa 95°C<br />
(temperatura di<br />
denaturazione)<br />
circa 55 °C<br />
(temperatura di<br />
annealing)<br />
circa 72°C<br />
(temperatura<br />
ottimale per la<br />
Taq)
L’operazione può essere<br />
ripetuta per diverse volte.<br />
Ciò consente anche alle<br />
catene sintetizzate ad ogni<br />
ciclo di fare da stampo in<br />
quello successivo, dando<br />
così un’amplificazione<br />
esponenziale.<br />
Considerando che parte<br />
dell’enzima e dei<br />
deossinucleotidi trifosfato<br />
si degradano ad ogni ciclo,<br />
il numero massimo di cicli<br />
è limitato (generalmente<br />
Si noti che, nei primi cicli, il prodotto è parzialmente<br />
eterogeneo. Quando i primer si legano al materiale di<br />
partenza o a prodotto di amplificazione che in tutti i cicli sia<br />
stato ottenuto a partire dallo stesso primer tra i due presenti,<br />
la polimerasi può continuare indefinitamente e la lunghezza<br />
per più cicli di seguito con lo stesso primer tra i due presenti<br />
della catena prodotta dipenderà prevalentemente dal tempo<br />
che la polimerasi ha a disposizione per la sua reazione.<br />
Se invece il primer si lega ad una catena derivante dalla<br />
polimerizzazione con l’altro primer, il tratto amplificato finale<br />
sarà quello delimitato dai due primer.<br />
È estremamente improbabile che un segmento venga<br />
amplificato. Dopo pochi cicli vengono quindi a prevalere i<br />
segmenti “corti”, di lunghezza ben definita. <strong>Il</strong> prodotto finale è<br />
pressochè puro.<br />
Alla fine dei cicli, si lascia il DNA alla temperatura ottimale per<br />
la sua rinaturazione, per cui i filamenti complementari,<br />
presenti in ugual quantità, si appaiano dando luogo a doppie<br />
eliche.<br />
Si noti anche che, durante l’intervallo alla temperatura di<br />
annealing, i filamenti di DNA potrebbero anche combinarsi<br />
con i filamenti complementari pittosto che con i primers. I<br />
primi ovviamente non potrebbero costituire uno stampo per la<br />
DNA polimerasi. II numero di doppie eliche complete che si<br />
formano durante l’annealing è tuttavia minimo, dato che i<br />
primer vengono aggiunti in grande eccesso. Solo dopo alcuni<br />
cicli i primer si esauriscono.
Le tecniche del DNA ricombinante e della PCR possono essere<br />
integrate: I primer per la PCR devono essere complementari ai<br />
due estremi del DNA che interessa, ma possono anche<br />
contenere, a monte (posizione 3’) della regione complementare,<br />
una corta sequenza che non lo sia. Tale sequenza viene<br />
incorporata nel prodotto finale della PCR.<br />
5’ – GGGCCCTTTAAAATCGCA ATGTTAACGGGGCCCTTTAAAATC........TGTTTAAACCCGGGTTT TAA TTTAAACCCGGGTTT-3’<br />
3’ – CCCCGGGAAATTTTAGCGATACAATTGCCCCGGGAAATTTTAG.......AC.AAATTTGGGCCCAAAATT AAATTTGGGCCCAAA 5’<br />
3’ - CTTAAG<br />
PCR<br />
5’ – GGGCCCTTTAAAATCGCA ATGTTAACGGGGCCCTTTAAAATC........TGTTTAAACCCGGGTTTTAATTTAAACCCGGGTTT -3’<br />
TACAATTGCCCCGGGAAATTT<br />
TTTAAACCCGGGTTTTAA<br />
AAGCTT – 3’<br />
3’- CCCCGGGAAATTTTAGCGATACAATTGCCCCGGGAAATTTTAG........ ACAAATTTGGGCCCAAAATTTTTAAACCCGGGTTT - 5’<br />
In questo modo possono essere introdotti siti di restrizione,<br />
che possono venire impiegati per unire il prodotto PCR a<br />
altre molecole di DNA.
RT-PCR<br />
La RT-PCR (reverse transcriptase PCR) consente di<br />
trascrivere un filamento di RNA (generalmente mRNA) in DNA<br />
e amplificarlo. <strong>Il</strong> DNA che si ottiene prende il nome di cDNA.<br />
La RT-PCR ha due fondamentali applicazioni:<br />
1. Quantificare il livello di trascrizione di un gene, per valutare<br />
le risposte cellulari a particolari situazioni (nutrimenti, stress).<br />
Al limite si può analizzare l’intero trascrittoma, ovvero<br />
l’insieme dell’mRNA presente in un dato momento nella<br />
cellula (e quindi, indirettamente, della quantità di proteina<br />
espressa).<br />
2. Un secondo motivo per voler amplificare dell’mRNA<br />
anziché il gene da cui è stato trascritto si pone per gli<br />
eucarioti: il gene eucariotico contiene generalmente introni,<br />
che non verranno poi codificati. <strong>Il</strong> messaggio che verrà<br />
tradotto nella proteina risiede quindi nel mRNA. I batteri non<br />
sono in grado di operare lo splicing, per cui occorre fornire<br />
loro il “codice” che è già stato processato dagli eucarioti,<br />
ovvero l’mRNA maturo, trascritto in DNA mediante RT-PCR.
Non vi sono enzimi cellulari che consentano di amplificare il<br />
mRNA (né avrebbero senso, dato il ruolo che ricopre l’RNA<br />
nelle cellule). Si può però sfruttare l’enzima virale trascrittasi<br />
inversa, utilizzata dai retrovirus nel primo passaggio di<br />
integrazione del loro genoma a RNA in quello della cellula<br />
ospite. La trascrizione inversa è quindi un processo che non<br />
avviene normalmente negli organismi cellulari, né eucariotici<br />
né procariotici. Si tratta di un meccanismo attraverso il quale<br />
alcuni virus che presentano un genoma a RNA (retrovirus)<br />
possono “trascriverlo” a DNA in modo che possa essere<br />
integrato al genoma della cellula infettata.<br />
Dopo l’ingresso del virus nella cellula, la trascrittasi inversa<br />
usa l’RNA virale come stampo per sintetizzare una catena di<br />
DNA complementare, dando luogo ad una elica ibrida DNA-<br />
RNA. La catena di RNA virale viene poi degradata dalla<br />
RNasiH (un enzima virale facente parte della stessa catena<br />
polipeptidica della trascrittasi inversa). Infine, la trascrittasi<br />
inversa forma la catena complementare e quindi la doppia<br />
elica.<br />
Come le DNA polimerasi, la trascrittasi inversa necessita di<br />
un primer (ne occorre uno anziché due come nella normale<br />
PCR, perché l’RNA è a singolo filamento).
Una volta che il genoma virale si è integrato con il genoma ospite,<br />
le proteine virali codificate dal materiale genetico virale sono<br />
espresse dal sistema di trascrizione cellulare. Le proteine virali,<br />
insieme al suo genoma a RNA (quindi trascritto dalle RNA<br />
polimerasi della cellula sotto il controllo di una regolazione virale)<br />
possono riassemblarsi a dare nuovi virus.
Siccome le proteine virali sono poche, sono anche<br />
pochi i possibili bersagli molecolari per un’azione<br />
antivirale. La trascrittasi inversa è uno di questi. Gli<br />
inibitori della trascrittasi inversa sono i primi farmaci<br />
anti-HIV ad essere stati utilizzati.
Per poter trascrivere un filamento di RNA in DNA, la<br />
trascrittasi inversa necessita, come la taq polimerasi, di un<br />
primer.<br />
Nei procarioti e in generale quando si voglia amplificare un<br />
mRNA specifico si deve ricorrere a primer sintetici che<br />
abbiano sequenza complementare all’mRNA che interessa, in<br />
modo analogo a quanto visto per la PCR.<br />
Un’importante modificazione posttrascrizionale tipica degli<br />
eucarioti, la poliadenilazione in 3’, offre un modo per<br />
amplificare contemporaneamente tutti gli mRNA presenti<br />
impiegando come primer universale un poliT. La coda di poliA<br />
è anche sfruttata per separare gli mRNA dalle altre molecole<br />
presenti in un lisato cellulare.
Supponiamo di voler<br />
amplificare tutto il<br />
trascrittoma di cellule di un<br />
tessuto. Si estrae il mRNA<br />
maturo e lo purifica.<br />
L’mRNA viene a questo<br />
punto incubato con il primer<br />
poli T e con la trascrittasi<br />
inversa. Si forma una<br />
doppia elica ibrida<br />
DNA/RNA.<br />
Aggiungiamo l’enzima<br />
RNaseH, chè è in grado di<br />
idrolizzare il filamento di<br />
RNA, lasciando quello<br />
singolo di DNA. In<br />
alternativa, si può elevare il<br />
pH: l’RNA non è stabile<br />
all’idrolisi alcalina e quindi<br />
rimane solo il filamento di<br />
DNA.<br />
Se nella miscela di<br />
reazione è presente una<br />
DNA polimerasi e i 4<br />
deossinucleotidi trifosfato, i<br />
singoli filamenti di DNA<br />
possono generare le doppia<br />
eliche di DNA.
Se nell’ambiente di reazione è presente la taq polimerasi e i<br />
primers specifici per l’amplificazione di uno specifico gene, si<br />
può procedere direttamente all’amplificazione del cDNA per<br />
PCR.
Un passaggio in RT-PCR è indispensabile quando si voglia<br />
amplificare il DNA codificante per una proteina eucariotica e<br />
portarlo ad una forma “leggibile” dai procarioti, che non sono<br />
in grado di riconoscere e eliminare gli introni dall’mRNA.
Esempio: ottenimento di un cDNA del gene dell’insulina.
Sequenziamento del DNA<br />
<strong>Il</strong> metodo di elezione per il sequenziamento di catene di DNA<br />
fino a 500 bp è quello enzimatico, detto anche di Sanger. Si<br />
tratta di una reazione catalizzata da una DNA polimerasi su un<br />
singolo filamento condotta in presenza non solo dei quattro<br />
deossinucleotidi trifosfato, ma anche di loro derivati privi<br />
dell’ossidrile in 3’ (dideossinucleotidi), aggiunti in minima<br />
quantità.<br />
Per avere più materiale, si può condurre una PCR, ovviamente<br />
usando la Taq polimerasi come DNA polimerasi.
Affinchè la DNA polimerasi possa formare il filamento<br />
complementare, occorre fornire anche un primer, che individua<br />
l’inizio del tratto da sequenziare.<br />
Nelle prime versioni di questo metodo, si conducevano<br />
separatamente 4 reazioni di PCR, ognuna in presenza di un<br />
diverso nucleotide modificato. La duplicazione si blocca, per<br />
ognuna delle 4 reazioni, quando viene incorporato il nucleotide<br />
modificato: tali derivati possono essere aggiunti alla catena<br />
nascente dalla DNA polimerasi ma poi ne impediscono il<br />
proseguimento. <strong>Il</strong> loro inserimento è casuale, per cui si<br />
generano filamenti di lunghezza diversa, ciascuna terminante<br />
con il derivato terminatore.<br />
La proporzione di nucleotidi “normali” e modificati è tale per cui,<br />
statisticamente, si formeranno tutti i filamenti tronchi terminanti<br />
con un determinato nucleotide.<br />
n<br />
copie
Le catene sono tutte marcate radioattivamente (si aggiungono<br />
nucleotidi con un atomo radioattivo, o al primer o a una porzione<br />
dei nucleotidi trifosfato), per cui possono essere visualizzate<br />
mediante autoradiografia su un gel. Si utilizzano gel di<br />
poliacrilamide perché consentono di separare molecole che<br />
differiscono in lunghezza per un singolo nucleotide.
I 4 prodotti di PCR vengono fatti correre parallelamente su un<br />
gel. In questo modo è possibile ricostruire la sequenza completa<br />
del filamento complementare.
La variante più recente di questo metodo prevede che i quattro<br />
nucleotidi modificati leghino un label fluorescente, ognuno in<br />
grado di emettere luce di “colore” diverso. Con questo metodo<br />
è sufficiente una singola reazione di PCR in presenza di tutti e<br />
quattro i nucleotidi modificati. Le repliche parziali vengono<br />
separate mediante elettroforesi: il “colore” di ciascuna banda<br />
indicherà la natura del nucleotide terminate (A, T, G o C). In<br />
questo modo, si può ricostruire a l’intera sequenza.
Clonaggio<br />
<strong>Il</strong> clonaggio consiste nello sfruttare la capacità di cellule,<br />
generalmente batteriche, di ospitare DNA estraneo e di<br />
consentirne l’amplificazione insieme al proprio. Attraverso il<br />
clonaggio, possiamo quindi disporre di quantità illimitate di DNA<br />
di sequenza desiderata. <strong>Il</strong> clonaggio è quindi un’amplificazione in<br />
vivo di DNA ricombinante.<br />
Se lo si inserisse come tale in una cellula, il DNA non sarebbe<br />
replicato e si perderebbe immediatamente. Per poter essere<br />
mantenuto nelle cellule batteriche da una generazione all’altra, il<br />
frammento che vogliamo amplificare deve essere (a) integrato<br />
nel genoma cellulare oppure (b) essere inserito in una molecola<br />
di DNA extragenomico di cui facciano parte sequenze tali da<br />
consentirne la permanenza nella cellula e la replicazione. Nel<br />
secondo caso, di gran lunga più frequente, si parla di vettori di<br />
clonaggio. Quelli più utilizzati sono i plasmidi, piccoli cromosomi<br />
circolari accessori derivanti da varianti naturali normalmente<br />
presenti nelle cellule batteriche. I plasmidi permettono di clonare<br />
tratti di DNA corrispondenti a uno o pochi geni. Recentemente,<br />
con la possibilità di sequenziare interi genomi, si sono resi utili<br />
vettori di clonaggio più “capienti”, grazie ai quali è possibile<br />
clonare tratti di DNA molto più lunghi. I cromosomi artificiali<br />
batterici (BAC), i fagi e i cosmidi ne sono esempi.<br />
Per il clonaggio sono generalmente impiegati ceppi batterici da<br />
cui siano stati eliminati i sistemi di metilazione/restrizione, in<br />
modo che non interferiscano con il DNA che vogliamo inserire. Si<br />
noti che i vettori di clonaggio possono essere facilmente separati<br />
dalle altre componenti della cellula (compreso il suo genoma) e il<br />
frammento che vi abbiamo inserito può essere recuperato dal<br />
plasmide tagliandolo con gli stessi enzimi di restrizione con i<br />
quali lo abbiamo inserito.
I plasmidi sono elementi genetici accessori normalmente<br />
presenti nelle cellule procariotiche.<br />
• Contrariamente al cromosoma principale, i plasmidi sono<br />
normalmente presenti in più di una copia, spesso in numero<br />
variabile da cellula a cellula. Non sono generalmente<br />
indispensabili alla crescita della cellula in condizioni normali, ma<br />
codificano per proteine coinvolte nella resistenza ad antibiotici,<br />
proteine associate alla virulenza, ma anche enzimi che<br />
catalizzano particolari reazioni metaboliche.<br />
• I plasmidi sono entità genetiche relativamente indipendenti e<br />
possono spesso trasferirsi da cellula a cellula, anche tra specie<br />
diverse.
Type of plasmid<br />
Resistance<br />
Fertility<br />
Killer<br />
Degradative<br />
Virulence<br />
Gene functions<br />
Antibiotic resistance<br />
Conjugation and<br />
DNA transfer<br />
between bacteria<br />
Synthesis of toxins<br />
that kill other bacteria<br />
Enzymes for<br />
metabolism of<br />
unusual molecules<br />
Pathogenicity<br />
Examples<br />
Rbk of Escherichia<br />
coli and other<br />
bacteria<br />
F of E. coli<br />
Col of E. coli, for<br />
colicin production<br />
TOL of<br />
Pseudomonas<br />
putida, for toluene<br />
metabilism<br />
Ti of Agrobacterium<br />
tumefaciens,<br />
conferring the ability<br />
to cause crown gall<br />
disease on<br />
dicotyledonous<br />
plants
I plasmidi ricombinanti<br />
L’inserimento di plasmidi in batteri viene chiamato<br />
trasformazione ed è ottenuto trattando una cultura di batteri<br />
con agenti chimici o fisici che ne aumentano<br />
temporaneamente la permeabilità di membrana.
I plasmidi più comunemente usati come vettori di clonaggio<br />
hanno dimensioni variabili tra 2000 e 5000 bp e sono stati<br />
originariamente ottenuti da plasmidi naturali, tra cui, in<br />
particolare, il plasmide di E. coli ColE1. Alcuni possono<br />
replicarsi in più specie, altri sono più selettivi e possono<br />
replicarsi solo in una. Nella loro sequenza devono contenere<br />
almeno:<br />
1. Un’origine di replicazione (ori) affinché possano essere<br />
duplicati ad ogni ciclo cellulare. <strong>Il</strong> tipo di origine di<br />
duplicazione determina il numero medio di copie presenti nel<br />
batterio: si distinguono ori che danno un high copy number<br />
da quelle che danno un low copy number. I primi<br />
consentono di ottenere una quantità maggiore di DNA.<br />
L’origine di replicazione è spesso specie-specifica. Si può<br />
però modificare la sequenza del plasmide in modo che<br />
contenga siti ori specifici per più di un organismo.<br />
2. Un marker, o selettore: tra le funzioni svolte dai plasmidi in<br />
natura, vi è quella di veicolare un’attività antibiotica (vedi ad<br />
esempio le multiresistenze nelle infezioni nosocomiali).<br />
Codificano cioè per una o più proteine in grado di bloccare<br />
l’azione di un antibiotico (ad esempio, ampicillina,<br />
kanamicina, streptomicina). A livello molecolare, tale azione<br />
può essere di diverso tipo: ad esempio il gene di resistenza<br />
alla ampicillina codifica per un enzima, la β-lattamasi, che<br />
rompe l’antibiotico, inattivandolo. Si è pensato di sfruttare<br />
questa funzione dei plasmidi (che non è non l’unica in<br />
natura) per mantenere il plasmide nel batterio durante i vari<br />
cicli di duplicazione cellulare. Se si aggiunge l’antibiotico al<br />
mezzo di cultura, solo i batteri in cui il plasmide è presente<br />
sopravviveranno e si riprodurranno. In assenza di antibiotico<br />
il plasmide tenderebbe a perdersi. In un plasmide possono<br />
essere presenti geni di resistenza a più di un antibiotico.
La resistenza all’antibiotico è lo stratagemma che si usa per<br />
selezionare i cloni batterici in cui la trasformazione con il<br />
plasmide ricombinante abbia avuto successo (in genere, una<br />
minima parte). La cultura trattata con il plasmide viene “stesa”<br />
su un terreno solido (LB agar) in cui sia disciolto l’antibiotico al<br />
quale il plasmide conferisce resistenza. Le cellule nelle quali è<br />
presente potranno crescere come popolazione clonale (cellule<br />
identiche, derivate da una singola cellula) fino a formare<br />
colonie visibili ad occhio nudo. La colonia a questo punto può<br />
essere isolata e coltivata in terreni liquidi. <strong>Il</strong> plasmide può<br />
essere purificato dalla cultura batterica in modo semplice e<br />
veloce.
3. Un sito in cui siano contenuti dei siti di restrizione unici<br />
(single cutters), in modo da potervi inserire il frammento di<br />
DNA tagliato con gli stessi enzimi. Sarebbe sufficiente anche<br />
un solo sito di restrizione unico, ma molti plasmidi<br />
commerciali presentano un multiple cloning site, in modo<br />
che vi sia un’ampia scelta di enzimi di restrizione da usare<br />
(nota bene: la scelta dei siti di restrizione da impiegare per<br />
ottenere DNA ricombinante è ristretta dalla possibilità che<br />
tali enzimi taglino il frammento di DNA che interessa).<br />
<strong>Il</strong> MCS non è indispensabile al plasmide in sé ma è richiesto<br />
affinché lo si possa usare come vettore di clonaggio. Un<br />
MCS consente inoltre di clonare in serie più frammenti.
I BAC ricombinanti<br />
I BAC (cromosomi artificiali batterici) sono costrutti di DNA<br />
basati sul plasmide naturale della fertilità (F) di E. coli, più<br />
grande di altri plasmidi che si riscontrano in natura. Rispetto ai<br />
plasmidi, I BAC possono accomodare frammenti più lunghi di<br />
DNA (fino a 300 Kbp). Grazie a questa capienza, sono spesso<br />
impiegati nel seqenziamento di genomi.
I fagi ricombinanti<br />
I batteriofagi, o fagi, sono virus che infettano i batteri. <strong>Il</strong> loro<br />
materiale genetico, una volta entrato nella cellula può (a) integrarsi<br />
con quello della cellula ospite, di cui diventa parte integrante (via<br />
lisogenica) (b) moltiplicarsi in più copie: i geni virali vengono quindi<br />
espressi in grande quantità e diversi virus si assemblano,<br />
comportando la rottura della cellula (via litica). Si dicono temperati i<br />
fagi che vanno generalmente incontro a lisogenia, ma che sono<br />
occasionalmente in grado di iniziare un ciclo litico. <strong>Il</strong> batteriofago<br />
temperato più studiato è il batteriofago λ.<br />
L’integrazione del genoma virale è una ricombinazione non omologa<br />
(mediata da proteine che riconoscono siti specifici nel DNA), del tipo<br />
incontrato per molti virus.
Tanto il processo lisogenico che quello litico avvengono<br />
anche se, all’interno del genoma virale, inseriamo un<br />
frammento di DNA estraneo al fago. I fagi ricombinanti che si<br />
ottengono possono infettare cellule di E. coli come quelli<br />
naturali. Si può introdurre direttamente il genoma<br />
(trasfezione) o infettare le cellule con l’intero virus<br />
assemblato in vitro (infezione).<br />
trasfezione
<strong>Il</strong> genoma fagico ricombinante può essere introdotto nella cellula<br />
ospite per infezione, “impacchettandolo invitro con le proteine del<br />
capside.<br />
Affinché si possa avere l’impacchettamento in vitro del genoma<br />
fagico ricombinante a partire dalle proteine del capside, sono<br />
indispensabili le sequenze cos, situate normalmente alle<br />
estremità del genoma fagico. Tali sequenze sono riconosciute<br />
dalle proteine fagiche (codificate cioè da geni del genoma fagico)<br />
Nu1 e A, che ne mediano l’impaccamento nella “testa” del virus.<br />
Per un efficiente impacchettamento, è anche critica la lunghezza<br />
del DNA. Se è troppo corta (ad esempio non si è avuta<br />
l’inserzione del frammento esogeno), non si ha formazione di un<br />
fago attivo. Se è troppo lunga, l’impaccamento è impedito.<br />
infezione
La resa del processo di trasfezione/infezione è relativamente<br />
bassa, per cui solo poche cellule la subiranno. Incubando<br />
cellule di E. coli stratificate uniformemente su un terreno solido, si<br />
osserveranno delle placche (cellule lisate) che si originano da un<br />
singolo “focolaio” di infezione. I fagi che si trovano nella placca,<br />
derivando da un singolo fago, costituiranno una popolazione<br />
clonale (copie identiche del fago originario).<br />
I fagi di una placca potranno essere prelevati e usati per infettare<br />
una cultura in terreno liquido: si viene quindi ad ottenere un<br />
numero di fagi sufficiente per purificarne il DNA contenuto e, con<br />
esso, il nostro frammento.
I cosmidi ricombinanti<br />
I cosmidi sono plasmidi in cui<br />
sono stati inseriti siti cos, che<br />
consentono loro di impaccarsi -<br />
in vitro - nella testa di un<br />
batteriofago λ. Possono anche<br />
essere visti come batteriofagi cui<br />
sono stati tolti tutti i geni fagici,<br />
rimpiazzati con gli elementi tipici<br />
di un plasmide: ori, markers,<br />
MCS. Nella testa del virus, vi è<br />
quindi sufficiente spazio per un<br />
grosso inserto, fino a 45kb. Un<br />
inserto di tali dimensioni non<br />
potrebbe essere inserito in un<br />
fago λ (perché gran parte dello<br />
“spazio disponibile” è occupato<br />
dai geni fagici), ma neppure in un<br />
plasmide. In sostanza, con<br />
l’aggiunta dei siti cos, si<br />
consente l’introduzione del<br />
plasmide per infezione, un<br />
sistema più efficiente della<br />
trasformazione.<br />
Una volta entrato nella cellula, il<br />
cosmide si comporta come un<br />
normale plasmide (non possiede<br />
i geni del batteriofago per la<br />
ricombinazione, né per la sintesi<br />
delle proteine del capside).
Vector Type<br />
Plasmid<br />
λ phage<br />
Cosmid<br />
P1 phage<br />
BAC (bacterial artificial<br />
chromosome)<br />
YAC (yeast artificial chromosome)<br />
Cloned DNA (kb)<br />
20<br />
25<br />
45<br />
100<br />
300<br />
1000
Clonaggio di interi genomi:<br />
le librerie genomiche<br />
Tra le principali applicazioni del clonaggio vi è la possibilità di<br />
amplificare corti tratti di DNA ottenuti per frazionamento di<br />
molecole molto lunghe. I frammenti amplificati per clonaggio,<br />
disponibili in quantità pressoché illimitata, possono essere<br />
caratterizzati e sequenziati. Ad esempio, è possibile tagliare un<br />
grosso frammento di DNA con un singolo enzima di restrizione. Ne<br />
deriveranno frammenti più piccoli che possono essere inseriti in un<br />
plasmide tagliato con lo stesso enzima di restrizione.<br />
Si noti che i frammenti sono casuali e non rispecchiano<br />
necessariamente i limiti tra unità funzionali (geni, elementi di<br />
regolazione, enhancer, ecc).
I plasmidi possono essere trasformati in batteri a dare diversi<br />
cloni batterici, facilmente isolabili e amplificabili. L’insieme di<br />
questi cloni, che potenzialmente coprono tutto il DNA<br />
originario, prende il nome di libreria.<br />
Questa operazione potrà essere eventualmente ripetuta<br />
rompendo il DNA originario con altri enzimi di restrizione: si<br />
ottengono librerie ridondanti con frammenti in parte sovrapposti<br />
(contigs). L’analisi delle sequenze sovrapposte consente di<br />
risalire all’ordine in cui i frammenti si presentano nella<br />
molecola di DNA originaria (metodo “shotgun”) e, quindi, alla<br />
sua sequenza complessiva.
Al limite, si può costruire una vera e propria libreria genomica,<br />
un insieme di cloni batterici che nel loro complesso contengono<br />
tutto il DNA di un organismo. Per queste applicazioni, dovranno<br />
essere impiegati i vettori di clonaggio più “capienti” (fagi o<br />
BAC).<br />
Ad esempio, l’intero genoma umano è stato frazionato<br />
(Progetto Genoma Umano) in 393216 BACs.<br />
Queste librerie, eventualmente “sfoltite” per essere il meno<br />
ridondanti possibile, sono rese disponibili alla comunità<br />
scientifica.
Clonaggio di interi trascrittomi: le librerie di cDNA<br />
Le librerie genomiche non danno informazioni circa il diverso<br />
livello di espressione dei geni. Da questo punto di vista, è più<br />
informativa una libreria di cDNA, ottenuta mediante clonaggio del<br />
cDNA ottenuto per RT-PCR su tutti gli mRNA espressi da una<br />
cellula. Non si arriverà a clonare tutto il materiale genetico (non<br />
tutto è trascritto a mRNA), ma si ottengono informazioni, seppur<br />
indirettamente, sulle proteine espresse. Confrontando librerie<br />
genomiche con quelle di cDNA si possono avere informazioni<br />
sulla forza di promotori ed enhacer, sulla inducibilità di alcuni<br />
geni, sullo splicing, ecc.
Clonaggio di sequenze specifiche<br />
<strong>Il</strong> clonaggio può essere integrato con la PCR per isolare una<br />
sequenza ben definita ed eventualmente modificata. Inserendo<br />
siti di restrizione ai lati del segmento amplificato (usando<br />
opportuni primers), è possible inserirlo agevolmente in un<br />
vettore di clonaggio.<br />
Si noti che, se si mantiene il sito di restrizione grazie al quale si<br />
è inserito l’inserto nel plasmide, lo si potrà sempre recuperare<br />
e clonare, ad esempio, in un altro vettore (subclonaggio).
Espressione<br />
Un semplice clonaggio consente di isolare e caratterizzare un<br />
segmento di DNA, che può essere replicato a piacere in modo<br />
semplice ed efficace. Se il clonaggio viene condotto in<br />
appositi vettori, detti vettori di espressione, è possibile<br />
sfruttare non solo il sistema di duplicazione del DNA della<br />
cellula procariotica ospite, ma anche il suo sistema di<br />
trascrizione, ottenendo così la proteina codificata dal tratto di<br />
DNA (via mRNA). Se l’espressione avviene in un organismo<br />
diverso da quello in cui è stato isolato il gene si parla di<br />
espressione eterologa.<br />
Ciò è di straordinaria utilità, perché le proteine espresse e<br />
purificate possono trovare le più varie applicazioni. È inoltre<br />
possibile produrre di una proteina in quantità sufficiente per<br />
essere caratterizzata mediante tecniche biochimiche.<br />
Combinando questa tecnologia con quella della mutagenesi<br />
sito-specifica, è possibile esprimere e caratterizzare anche<br />
proteine mutanti.<br />
Nella grande maggioranza dei casi, la cellula ospite è<br />
batterica (E. coli). Le cellule batteriche non sono in grado di<br />
operare uno splicing sugli mRNA, per cui, volendo clonare ed<br />
esprimere una proteina eucariotica, dovremo utilizzare il<br />
cDNA ottenuto mediante RT-PCR dall’mRNA maturo estratto<br />
dalla cellula eucariotica. Se usassimo il prodotto PCR<br />
ottenuto dal genoma eucariotico, vi sarebbero inclusi anche<br />
gli introni, che la cellula batterica non saprebbe “interpretare”<br />
come tali.<br />
Rimane l’eventualità che la cellula procariotica non sia in<br />
grado di operare modificazioni post-traduzionali indispensabili<br />
alla funzione della proteina eucariotica. In questo caso è<br />
indispensabile esprimerla in cellule eucariotiche impiegando<br />
speciali vettori.
DNA<br />
Sequenza di regolazione<br />
della trascrizione (INIZIO<br />
trascrizione)<br />
mRNA<br />
Inizio trascrizione<br />
Proteina<br />
Espressione in procarioti<br />
Affinché un vettore di clonaggio sia anche di espressione, il<br />
frammento di DNA inserito deve essere un gene,<br />
comprendente le sequenze non codificanti di regolazione<br />
della sua espressione, sia a livello della trascrizione che<br />
della traduzione.<br />
Reg. trad.<br />
(INIZIO)<br />
Sequenza trascritta a RNA<br />
Fine trascrizione<br />
Inizio traduzione Fine traduzione<br />
Sequenza di regolazione<br />
della trascrizione (STOP<br />
trascrizione)<br />
Codone di stop
Sequenza di regolazione<br />
della trascrizione (INIZIO<br />
trascrizione)<br />
Promotore<br />
Inizio trascrizione<br />
Reg. trad.<br />
(INIZIO)<br />
RBS<br />
Sequenza trascritta a RNA<br />
Fine trascrizione<br />
Codone di stop<br />
Inizio traduzione Fine traduzione<br />
Sequenza di regolazione<br />
della trascrizione (STOP<br />
trascrizione)<br />
Sequenza di<br />
terminazione della<br />
trascrizione
Promotore<br />
La sintesi dell’RNA incomincia in corrispondenza di siti<br />
specifici sullo stampo di DNA: la RNA polimerasi si lega<br />
ad una sequenza di basi detta promotore.<br />
Nei procarioti, i promotori sono sequenze di DNA di circa<br />
40 basi localizzate immediatamente a monte del gene. Un<br />
tipico promotore batterico consiste di due sequenze di<br />
consenso (sequenze comuni a tutti i promotori): la<br />
sequenza TTGACA centrata in posizione -35 (riferito alla<br />
posizione di inizio della trascrizione) e quella TATAAT in<br />
posizione -10.
Ribosome binding site<br />
La sintesi proteica nei procarioti<br />
consiste di tre fasi: inizio,<br />
elongazione o allungamento e<br />
terminazione.<br />
L’inizio è innescato dal legame<br />
della subunità piccola, insieme<br />
al fattore IF-3, all’RBS. <strong>Il</strong><br />
legame con l’RBS consente il<br />
corretto posizionamento dei siti<br />
attivi del ribosoma sul codone di<br />
inizio dell’ mRNA.<br />
<strong>Il</strong> codone di inizio per i geni<br />
batterici è generalmente AUG, il<br />
codone per la metionina.<br />
A questo punto il tRNAi – met<br />
(formilata) si lega al suo<br />
codone.<br />
Infine, la subunità maggiore si<br />
lega alla subunità minore<br />
formando il complesso di<br />
iniziazione.<br />
Quindi: sia la sequenza RBS<br />
che il codone AUG definiscono<br />
il reading frame
<strong>Il</strong> plasmide contenente gli elementi genetici accessori necessari<br />
per la trascrizione (oltre a quelli necessari per la selezione e per<br />
la replicazione) può essere trasformato in una cellula<br />
procariotica come un plasmide di clonaggio. Come vettori di<br />
espressione per procarioti, è sufficiente utilizzare i plasmidi<br />
perché un gene è di dimensioni ridotte e non richiede vettori più<br />
capienti, come BAC o cosmidi.
Ciascun batterio possiede diversi tipi di siti promotori (siti cui si<br />
lega la RNA polimerasi), che potrebbero essere posti a monte<br />
di un gene plasmidico per regolarne la trascrizione. Tra i<br />
possibili siti promotori presenti nel genoma di E. coli, ci si è<br />
focalizzati su quelli che controllano geni inducibili. È infatti utile<br />
poter indurre l’espressione di una proteina mediante induttori<br />
(piccole molecole, possibilmente stabili) al momento<br />
desiderato, ad esempio quando la cultura ha raggiunto un certo<br />
livello di crescita, dato che la sovraespressione di una proteina<br />
esogena tende a rallentarla.<br />
Uno dei sistemi più semplici consiste nell’impiegare il<br />
promotore per l’operone del lattosio (o lac). Invece del lattosio,<br />
l’induttore naturale, si impiega un suo analogo sintetico più<br />
stabile, l’IPTG.
Le proteine repressore interagiscono direttamente con il DNA in<br />
corrispondenza di una sequenza specifica: quando vi si legano,<br />
impediscono alla RNA polimerasi di proseguire la trascrizione.<br />
L’associazione e la dissociazione di queste proteine regolatrici<br />
dal DNA è a sua volta regolata da piccole molecole, che<br />
legandosi a siti allosterici determinano modificazioni<br />
conformazionali tali da modificare le proprietà di legame della<br />
proteina al DNA.<br />
Le molecole sono spesso metaboliti o loro derivati: segnalano<br />
quindi al sistema di trascrizione quando debbano essere<br />
espresse proteine in risposta a una mutata situazione<br />
ambientale.<br />
L’esempio meglio studiato è quello del repressore lac. In<br />
presenza del suo ligando (l’allolattosio) il repressore si stacca<br />
dalla sequenza di regolazione
Come il lattosio, l’IPTG si lega alla proteina lacI,<br />
inducendone la dissociazione dal DNA e quindi<br />
consentendo alla RNA polimerasi di trascrivere il gene<br />
immediatamente a valle (che normalmente, nei batteri, è<br />
l’operone lac, ma che noi possiamo sostituire con il gene<br />
che ci interessa).
Per ottenere rese ancora più alte si può ricorrere ad un<br />
duplice sistema di amplificazione basato su un promotore<br />
virale, il T7, riconosciuto dalla RNA polimerasi virale T7.<br />
Vi sono ceppi ingegnerizzati di E. coli in cui il gene per la<br />
RNA polimerasi T7 viene posto sotto il controllo del<br />
promotore lac (quindi inducibile dall’IPTG). Una volta che ne<br />
è indotta l’espressione, la RNA polimerasi potrà a questo<br />
punto trascrivere il gene di interesse posto nel plasmide<br />
sotto il controllo del promotore virale T7. La produzione di<br />
polimerasi T7 determina un’enorme espressione dei geni<br />
dipendenti dal suo promotore (T7, appunto).
Se si mette a monte del promotore T7 anche un promotore<br />
lac, rendiamo ancora più stringente l’induzione: l’IPTG<br />
rimuoverà contemporaneamente la repressione sul gene<br />
plasmidico e sulla RNA polimerasi T7. Un gene per il<br />
repressore lac, lacI, può essere inserito nel plasmide per<br />
aumentarne la concentrazione all’interno della cellula, in<br />
modo che l’espressione sia completamente soppressa in<br />
assenza di induttore.<br />
Questi stratagemmi per rendere il più possibile stringente<br />
l’induzione sono motivati dal fatto che, a volte, la proteina<br />
sovraespressa in grandi quantità è tossica per la cellula. <strong>Il</strong><br />
controllo sui tempi di espressione diventa quindi cruciale per<br />
ottimizzare le condizioni di espressione.
Mutagenesi sito specifica
Espressione in eucarioti<br />
La maggior parte delle proteine eucariotiche può essere<br />
espresso in cellule procariotiche (via cDNA), ma alcune<br />
richiedono modificazioni post-traduzionali che i procarioti<br />
non riescono a compiere. In questi casi occorre quindi<br />
esprimere le proteine in sistemi eucariotici.<br />
Siccome la maggior parte degli eucarioti non tollera DNA<br />
extracromosomale (tipo plasmidi), il DNA che si vuole<br />
esprimere deve essere inserito, con gli elementi di<br />
regolazione riconosciuti dalla cellula ospite, nel DNA<br />
genomico.<br />
<strong>Il</strong> DNA può essere microiniettato in cellule animali. il tasso di<br />
successo è molto basso, perché un’inserzione spontanea<br />
nel genoma è molto improbabile.
Si ha maggiore efficienza utilizzando vettori di espressione<br />
per eucarioti, generalmente derivati da virus, dei quali si<br />
sfrutta la capacità, mediante ricombinazione, di inserire<br />
DNA nel genoma della cellula ospite (come i fagi nella via<br />
lisogena).<br />
Principi simili a quelli visti per i fagi possono essere ad<br />
esempio applicati al baculovirus, in grado di infettare cellule<br />
di insetto (che possono poi essere mantenute in cultura).<br />
Per ciascun tipo di eucariote si è sviluppato un set<br />
appropriato di vettori.
In genere si preferisce<br />
usare cellule eucariotiche<br />
in cultura, perché è più<br />
facile recuparare la<br />
proteina espressa.<br />
Un’eccezione è data da<br />
tessuti di animali dai quali<br />
la proteina è secreta e<br />
facilmente recuperabile.<br />
Ad esempio,<br />
un’importante farmaco<br />
proteico, l’attivatore<br />
tissutale del<br />
plasminogeno, viene<br />
secreto nel latte di pecore<br />
transgeniche. <strong>Il</strong> gene per<br />
l’attivatore tissutale del<br />
plasminogeno è in realtà<br />
presente in tutte le cellule<br />
ma è posto sotto il<br />
controllo del gene per la<br />
β-lattoglobulina, una<br />
proteina sintetizzata solo<br />
dalla ghiandola<br />
mammaria.