

Anforderungsformular Molekulargenetik mit Einwilligungsnachweis
Anforderungsformular Molekulargenetik mit Einwilligungsnachweis
Anforderungsformular Molekulargenetik mit Einwilligungsnachweis

Sie wollen auch ein ePaper? Erhöhen Sie die Reichweite Ihrer Titel.
YUMPU macht aus Druck-PDFs automatisch weboptimierte ePaper, die Google liebt.
Version: 20.08.2013<br />
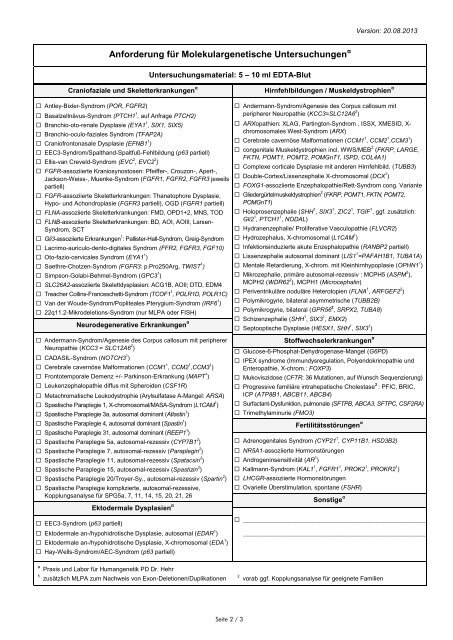
Anforderung für Molekulargenetische Untersuchungen α<br />
Untersuchungsmaterial: 5 – 10 ml EDTA-Blut<br />
Craniofaziale und Skeletterkrankungen α<br />
Antley-Bixler-Syndrom (POR, FGFR2)<br />
Basalzellnävus-Syndrom (PTCH1 1 , auf Anfrage PTCH2)<br />
Branchio-oto-renale Dysplasie (EYA1 1 , SIX1, SIX5)<br />
Branchio-oculo-faziales Syndrom (TFAP2A)<br />
Craniofrontonasale Dysplasie (EFNB1 1 )<br />
EEC3-Syndrom/Spalthand-Spaltfuß-Fehlbildung (p63 partiell)<br />
Ellis-van Creveld-Syndrom (EVC 2 , EVC2 2 )<br />
FGFR-assoziierte Kraniosynostosen: Pfeiffer-, Crouzon-, Apert-,<br />
Jackson-Weiss-, Muenke-Syndrom (FGFR1, FGFR2, FGFR3 jeweils<br />
partiell)<br />
FGFR-assoziierte Skeletterkrankungen: Thanatophore Dysplasie,<br />
Hypo- und Achondroplasie (FGFR3 partiell), OGD (FGFR1 partiell)<br />
FLNA-assoziierte Skeletterkrankungen: FMD, OPD1+2, MNS, TOD<br />
FLNB-assoziierte Skeletterkrankungen: BD, AOI, AOIII, Larsen-<br />
Syndrom, SCT<br />
Gli3-assoziierte Erkrankungen 1 : Pallister-Hall-Syndrom, Greig-Syndrom<br />
Lacrimo-auriculo-dento-digitales Syndrom (FFR2, FGFR3, FGF10)<br />
Oto-fazio-cervicales Syndrom (EYA1 1 )<br />
Saethre-Chotzen-Syndrom (FGFR3: p.Pro250Arg, TWIST 1 )<br />
Simpson-Golabi-Behmel-Syndrom (GPC3 1 )<br />
SLC26A2-assoziierte Skelettdysplasien: ACG1B, AOII; DTD, EDM4<br />
Treacher Collins-Franceschetti-Syndrom (TCOF1 1 , POLR1D, POLR1C)<br />
Van der Woude-Syndrom/Popliteales Pterygium-Syndrom (IRF6 1 )<br />
22q11.2-Mikrodeletions-Syndrom (nur MLPA oder FISH)<br />
Neurodegenerative Erkrankungen α<br />
Andermann-Syndrom/Agenesie des Corpus callosum <strong>mit</strong> peripherer<br />
Neuropathie (KCC3 = SLC12A6 2 )<br />
CADASIL-Syndrom (NOTCH3 1 )<br />
Cerebrale cavernöse Malformationen (CCM1 1 , CCM2 1 ,CCM3 1 )<br />
Frontotemporale Demenz +/- Parkinson-Erkrankung (MAPT 1 )<br />
Leukenzephalopathie diffus <strong>mit</strong> Spheroiden (CSF1R)<br />
Metachromatische Leukodystrophie (Arylsulfatase A-Mangel: ARSA)<br />
Spastische Paraplegie 1, X-chromosomal/MASA-Syndrom (L1CAM 1 )<br />
Spastische Paraplegie 3a, autosomal dominant (Atlastin 1 )<br />
Spastische Paraplegie 4, autosomal dominant (Spastin 1 )<br />
Spastische Paraplegie 31, autosomal dominant (REEP1 1 )<br />
Spastische Paraplegie 5a, autosomal-rezessiv (CYP7B1 2 )<br />
Spastische Paraplegie 7, autosomal-rezessiv (Paraplegin 2 )<br />
Spastische Paraplegie 11, autosomal-rezessiv (Spatacsin 2 )<br />
Spastische Paraplegie 15, autosomal-rezessiv (Spastizin 2 )<br />
Spastische Paraplegie 20/Troyer-Sy., autosomal-rezessiv (Spartin 2 )<br />
Spastische Paraplegie komplizierte, autosomal-rezessive,<br />
Kopplungsanalyse für SPG5a, 7, 11, 14, 15, 20, 21, 26<br />
EEC3-Syndrom (p63 partiell)<br />
Ektodermale Dysplasien α<br />
Ektodermale an-/hypohidrotische Dysplasie, autosomal (EDAR 1 )<br />
Ektodermale an-/hypohidrotische Dysplasie, X-chromosomal (EDA 1 )<br />
Hay-Wells-Syndrom/AEC-Syndrom (p63 partiell)<br />
Hirnfehlbildungen / Muskeldystrophien α<br />
Andermann-Syndrom/Agenesie des Corpus callosum <strong>mit</strong><br />
peripherer Neuropathie (KCC3=SLC12A6 2 )<br />
ARXopathien: XLAG, Partington-Syndrom , ISSX, XMESID, X-<br />
chromosomales West-Syndrom (ARX)<br />
Cerebrale cavernöse Malformationen (CCM1 1 , CCM2 1 ,CCM3 1 )<br />
congenitale Muskeldystrophien incl. WWS/MEB 2 (FKRP, LARGE,<br />
FKTN, POMT1, POMT2, POMGnT1, ISPD, COL4A1)<br />
Complexe corticale Dysplasie <strong>mit</strong> anderen Hirnfehlbild. (TUBB3)<br />
Double-Cortex/Lissenzephalie X-chromosomal (DCX 1 )<br />
FOXG1-assoziierte Enzephalopathie/Rett-Syndrom cong. Variante<br />
Gliedergürtelmuskeldystrophien 2 (FKRP, POMT1, FKTN, POMT2,<br />
POMGnT1)<br />
Holoprosenzephalie (SHH 1 , SIX3 1 , ZIC2 1 , TGIF 1 , ggf. zusätzlich:<br />
Gli2 1 , PTCH1 1 , NODAL)<br />
Hydranenzephalie/ Proliferative Vasculopathie (FLVCR2)<br />
Hydrozephalus, X-chromosomal (L1CAM 1 )<br />
Infektionsinduzierte akute Enzephalopathie (RANBP2 partiell)<br />
Lissenzephalie autosomal dominant (LIS1 1 =PAFAH1B1, TUBA1A)<br />
Mentale Retardierung, X-chrom. <strong>mit</strong> Kleinhirnhypoplasie (OPHN1 1 )<br />
Mikrozephalie, primäre autosomal-rezessiv : MCPH5 (ASPM 2 ),<br />
MCPH2 (WDR62 2 ), MCPH1 (Microcephalin)<br />
Periventrikuläre noduläre Heterotopien (FLNA 1 , ARFGEF2 2 )<br />
Polymikrogyrie, bilateral asymmetrische (TUBB2B)<br />
Polymikrogyrie, bilateral (GPR56 2 , SRPX2, TUBA8)<br />
Schizenzephalie (SHH 1 , SIX3 1 , EMX2)<br />
Septooptische Dysplasie (HESX1, SHH 1 , SIX3 1 )<br />
Stoffwechselerkrankungen α<br />
Glucose-6-Phosphat-Dehydrogenase-Mangel (G6PD)<br />
IPEX syndrome (Immundysregulation, Polyendokrinopathie und<br />
Enteropathie, X-chrom.: FOXP3)<br />
Mukoviszidose (CFTR: 36 Mutationen, auf Wunsch Sequenzierung)<br />
Progressive familiäre intrahepatische Cholestase 2 : PFIC, BRIC,<br />
ICP (ATP8B1, ABCB11, ABCB4)<br />
Surfactant-Dysfunktion, pulmonale (SFTPB, ABCA3, SFTPC, CSF2RA)<br />
Trimethylaminurie (FMO3)<br />
Fertilitätsstörungen α<br />
Adrenogenitales Syndrom (CYP21 1 , CYP11B1, HSD3B2)<br />
NR5A1-assoziierte Hormonstörungen<br />
Androgeninsensitivität (AR 1 )<br />
Kallmann-Syndrom (KAL1 1 , FGFR1 1 , PROK2 1 , PROKR2 1 )<br />
LHCGR-assoziierte Hormonstörungen<br />
Ovarielle Überstimulation, spontane (FSHR)<br />
Sonstige α<br />
______________________________________________________<br />
______________________________________________________<br />
α<br />
Praxis und Labor für Humangenetik PD Dr. Hehr<br />
1 zusätzlich MLPA zum Nachweis von Exon-Deletionen/Duplikationen<br />
2 vorab ggf. Kopplungsanalyse für geeignete Familien<br />
Seite 2 / 3

