

Crystallography and Lectin Structure Database - CNRS
Crystallography and Lectin Structure Database - CNRS
Crystallography and Lectin Structure Database - CNRS

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
18 U. Krengel <strong>and</strong> A. Imberty<br />
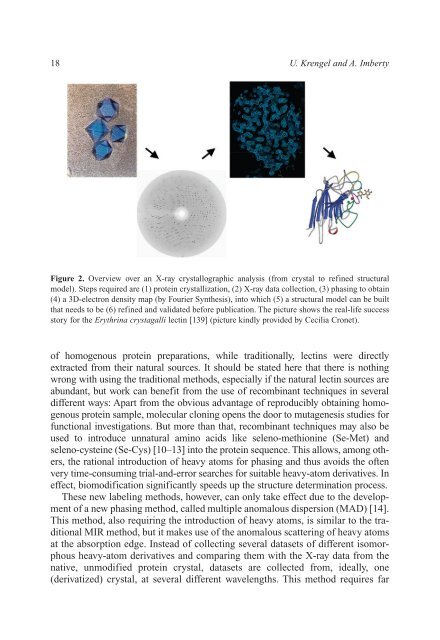
Figure 2. Overview over an X-ray crystallographic analysis (from crystal to refined structural<br />
model). Steps required are (1) protein crystallization, (2) X-ray data collection, (3) phasing to obtain<br />
(4) a 3D-electron density map (by Fourier Synthesis), into which (5) a structural model can be built<br />
that needs to be (6) refined <strong>and</strong> validated before publication. The picture shows the real-life success<br />
story for the Erythrina crystagalli lectin [139] (picture kindly provided by Cecilia Cronet).<br />
of homogenous protein preparations, while traditionally, lectins were directly<br />
extracted from their natural sources. It should be stated here that there is nothing<br />
wrong with using the traditional methods, especially if the natural lectin sources are<br />
abundant, but work can benefit from the use of recombinant techniques in several<br />
different ways: Apart from the obvious advantage of reproducibly obtaining homogenous<br />
protein sample, molecular cloning opens the door to mutagenesis studies for<br />
functional investigations. But more than that, recombinant techniques may also be<br />
used to introduce unnatural amino acids like seleno-methionine (Se-Met) <strong>and</strong><br />
seleno-cysteine (Se-Cys) [10–13] into the protein sequence. This allows, among others,<br />
the rational introduction of heavy atoms for phasing <strong>and</strong> thus avoids the often<br />
very time-consuming trial-<strong>and</strong>-error searches for suitable heavy-atom derivatives. In<br />
effect, biomodification significantly speeds up the structure determination process.<br />
These new labeling methods, however, can only take effect due to the development<br />
of a new phasing method, called multiple anomalous dispersion (MAD) [14].<br />
This method, also requiring the introduction of heavy atoms, is similar to the traditional<br />
MIR method, but it makes use of the anomalous scattering of heavy atoms<br />
at the absorption edge. Instead of collecting several datasets of different isomorphous<br />
heavy-atom derivatives <strong>and</strong> comparing them with the X-ray data from the<br />
native, unmodified protein crystal, datasets are collected from, ideally, one<br />
(derivatized) crystal, at several different wavelengths. This method requires far













