

The real structure of Na3BiO4 by electron ... - Columbia University
The real structure of Na3BiO4 by electron ... - Columbia University
The real structure of Na3BiO4 by electron ... - Columbia University

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Real <strong>structure</strong> <strong>of</strong> <strong>Na3BiO4</strong><br />
coordination sphere <strong>of</strong> the oxygen anion can best be described<br />
as a slightly distorted octahedron formed <strong>by</strong> sodium<br />
and bismuth.<br />
Due to the long coherence length <strong>of</strong> X-rays used, the<br />
average crystal <strong>structure</strong> <strong>of</strong> b-<strong>Na3BiO4</strong> was analyzed, resulting<br />
in a model with sodium und bismuth cations occupying<br />
almost statistically the same atomic positions. Because<br />
<strong>of</strong> the apparent diffuse scattering, the local order at<br />
the atomic level was studied <strong>by</strong> means <strong>of</strong> pair distribution<br />
function analysis.<br />
Pair distribution function analysis<br />
<strong>The</strong> <strong>real</strong>-space pair distribution function (PDF), G(r),<br />
gives the probability <strong>of</strong> finding pairs <strong>of</strong> atoms separated<br />
<strong>by</strong> distance r, and there<strong>by</strong> comprises peaks corresponding<br />
to all discrete interatomic distances. <strong>The</strong> experimental<br />
PDF is a direct Fourier transform <strong>of</strong> the total scattering<br />
<strong>structure</strong> function S(Q), the corrected, normalized intensity,<br />
from powder scattering data given <strong>by</strong><br />
where<br />
GðrÞ ¼ 2<br />
p<br />
ð1<br />
0<br />
Q½SðQÞ 1Š sin Qr dQ ;<br />
Q ¼ 4p<br />
sin q<br />
l<br />
is the magnitude <strong>of</strong> the scattering vector. Unlike crystallographic<br />
techniques, the PDF incorporates both Bragg and<br />
diffuse scattering intensities resulting in local structural information<br />
[44, 45]. Its high <strong>real</strong>-space resolution is ensured<br />
<strong>by</strong> measurement <strong>of</strong> scattering intensities over an extended<br />
Q range using short wavelength X-rays or<br />
neutrons.<br />
For the room-temperature data considered here, transformation<br />
<strong>of</strong> the FðQÞ ¼QðSðQÞ 1Þ; to a Qmax <strong>of</strong><br />
25.0 A 1 was found to be optimal. <strong>The</strong>re are basically two<br />
considerations. <strong>The</strong> first is to have sufficient Qmax to avoid<br />
large termination effects; the second is to reasonably minimize<br />
the noise level due to statistical fluctuations as the<br />
signal-to-noise ratio decreases with increasing Q. We<br />
found that Qmax <strong>of</strong> 25.0 A 1 has significantly lower noise<br />
level without losing useful structural information, i.e. no<br />
significant change <strong>of</strong> PDF peaks.<br />
<strong>The</strong> experimental PDF with Qmax 25.0 A 1 was refined<br />
within the crystallographic model <strong>of</strong> b-<strong>Na3BiO4</strong> as described<br />
in the chapter above. <strong>The</strong> constraints <strong>of</strong> space<br />
group R3m were maintained. Lattice parameters, thermal<br />
displacement parameters, and some experimental factors<br />
were refined. <strong>The</strong> occupancy <strong>of</strong> the atoms on each site<br />
was fixed according to the values (s<strong>of</strong> Rietveld) given in<br />
Table 2. We obtained lattice parameters <strong>of</strong> a ¼ b ¼<br />
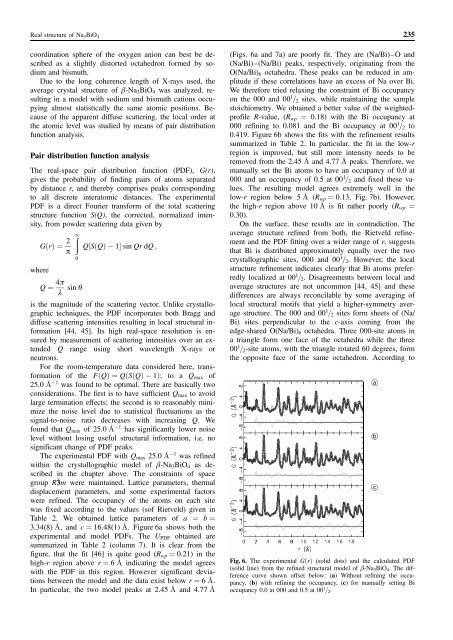
3.34(8) A, and c ¼ 16.48(1) A. Figure 6a shows both the<br />
experimental and model PDFs. <strong>The</strong> UPDF obtained are<br />
summarized in Table 2 (column 7). It is clear from the<br />
figure, that the fit [46] is quite good (Rwp ¼ 0.21) in the<br />
high-r region above r ¼ 6 A indicating the model agrees<br />
with the PDF in this region. However significant deviations<br />
between the model and the data exist below r ¼ 6 A.<br />
In particular, the two model peaks at 2.45 A and 4.77 A<br />
235<br />
(Figs. 6a and 7a) are poorly fit. <strong>The</strong>y are (Na/Bi)–O and<br />
(Na/Bi)–(Na/Bi) peaks, respectively, originating from the<br />
O(Na/Bi)6 octahedra. <strong>The</strong>se peaks can be reduced in amplitude<br />
if these correlations have an excess <strong>of</strong> Na over Bi.<br />
We therefore tried relaxing the constraint <strong>of</strong> Bi occupancy<br />
on the 000 and 00 1 =2 sites, while maintaining the sample<br />
stoichiometry. We obtained a better value <strong>of</strong> the weightedpr<strong>of</strong>ile<br />
R-value, (Rwp ¼ 0.18) with the Bi occupancy at<br />
000 refining to 0.081 and the Bi occupancy at 00 1 =2 to<br />
0.419. Figure 6b shows the fits with the refinement results<br />
summarized in Table 2. In particular, the fit in the low-r<br />
region is improved, but still more intensity needs to be<br />
removed from the 2.45 A and 4.77 A peaks. <strong>The</strong>refore, we<br />
manually set the Bi atoms to have an occupancy <strong>of</strong> 0.0 at<br />
000 and an occupancy <strong>of</strong> 0.5 at 00 1 =2 and fixed these values.<br />
<strong>The</strong> resulting model agrees extremely well in the<br />
low-r region below 5 A (Rwp ¼ 0.13, Fig. 7b). However,<br />
the high-r region above 10 A is fit rather poorly (Rwp ¼<br />
0.30).<br />
On the surface, these results are in contradiction. <strong>The</strong><br />
average <strong>structure</strong> refined from both, the Rietveld refinement<br />
and the PDF fitting over a wider range <strong>of</strong> r, suggests<br />
that Bi is distributed approximately equally over the two<br />
crystallographic sites, 000 and 00 1 =2. However, the local<br />
<strong>structure</strong> refinement indicates clearly that Bi atoms preferredly<br />
localized at 00 1 =2. Disagreements between local and<br />
average <strong>structure</strong>s are not uncommon [44, 45] and these<br />
differences are always reconcilable <strong>by</strong> some averaging <strong>of</strong><br />
local structural motifs that yield a higher-symmetry average<br />
<strong>structure</strong>. <strong>The</strong> 000 and 00 1 =2 sites form sheets <strong>of</strong> (Na/<br />
Bi) sites perpendicular to the c-axis coming from the<br />
edge-shared O(Na/Bi)6 octahedra. Three 000-site atoms in<br />
a triangle form one face <strong>of</strong> the octahedra while the three<br />
00 1 =2-site atoms, with the triangle rotated 60 degrees, form<br />
the opposite face <strong>of</strong> the same octahedron. According to<br />
Fig. 6. <strong>The</strong> experimental GðrÞ (solid dots) and the calculated PDF<br />
(solid line) from the refined structural model <strong>of</strong> b-<strong>Na3BiO4</strong>. <strong>The</strong> difference<br />
curve shown <strong>of</strong>fset below: (a) Without refining the occupancy,<br />
(b) with refining the occupancy, (c) for manually setting Bi<br />
occupancy 0.0 at 000 and 0.5 at 00 1 =2.<br />
a<br />
b<br />
c