Topic 2 lecture note..
Topic 2 lecture note..
Topic 2 lecture note..

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Cell adhesion: Roles in lymphocyte recirculation,<br />
trafficking, and microenvironment-specific homing<br />
A/Professor Geoff Krissansen Ext 86280<br />
Cell to cell, and cell to matrix interactions are fundamental biological processes which<br />
regulate cell motility, migration, localisation, differentiation, and proliferation in all<br />
multicellular organisms. In this course we will define the families of cell adhesion<br />
molecules which sit on the outside of cells enabling cells to communicate with one<br />
another, and to respond to the immediate microenvironment. Brief descriptions of the<br />
structures, functions, and expression of various cell adhesion molecules, including the<br />
Integrins, Ig CAMs, Cadherins, Selectins, Mucins, and extracellular matrix molecules.<br />
The "area code" molecules, in particular the chemokines, that direct leukocyte traffic, and<br />
microenvironment-specific lymphocyte homing will be analysed.<br />
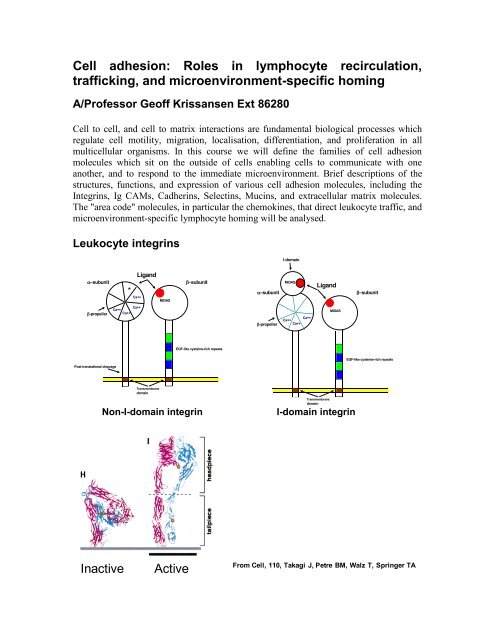
Leukocyte integrins<br />
I-domain<br />
subunit<br />
Ligand<br />
*<br />
Ca++<br />
MIDAS<br />
subunit<br />
subunit<br />
MIDAS<br />
Ligand<br />
subunit<br />
-propeller<br />
Ca++<br />
Ca++<br />
Ca++<br />
-propeller<br />
Ca++<br />
Ca++<br />
Ca++<br />
MIDAS<br />
EGF-like cysteine-rich repeats<br />
EGF-like cysteine-rich repeats<br />
Post-translational cleavage<br />
Transmembrane<br />
domain<br />
Non-I-domain integrin<br />
Transmembrane<br />
domain<br />
I-domain integrin<br />
Inactive<br />
Active<br />
From Cell, 110, Takagi J, Petre BM, Walz T, Springer TA
L, M, X, D, and E integrins are major receptors of the immune system<br />
A subgroup of 2 integrins<br />
The four -subunits of this subgroup assemble with the integrin 2-subunit to generate<br />
the integrins L2 (LFA-1, CD11a/CD18), M2 (Mac-1, Mo-1, CR3, CD11b/CD18),<br />
X2 (p150,95, CR4, CD11c/CD18), and D2 (CD11d/CD18) (Springer, 1995). They<br />
are restricted to leukocytes, where L2 is expressed by most leukocytes, and M2 and<br />
X2 are on monocytes/macrophages, granulocytes, large granular lymphocytes, and a<br />
subpopulation of immature B cells. X2 is also present on some activated T cells.<br />
L2: L2 mediates the adhesion of leukocytes to the intercellular adhesion molecules<br />
ICAM-1, ICAM-2, and ICAM-3, which are inducibly or constitutively expressed on<br />
many cell types. It participates in leukocyte transendothelial migration, recirculation,<br />
homing, and localization to inflammatory sites. L2 plays a key role in antigenpresentation,<br />
T-cell costimulation, the cytotoxicity of T cells, delayed-type<br />
hypersensitivity, and endotoxin shock.<br />
Leukocytes from mice lacking L display defects in in vitro homotypic aggregation, in<br />
proliferation in mixed lymphocyte reactions, and in response to mitogen. In vivo, host-vsgraft<br />
reaction toward injected allogeneic cells is also reduced. Neutrophils and activated<br />
T lymphocytes are unable to cross endothelial cell monolayers in response to a<br />
chemokine gradient. The trafficking of lymphocytes to peripheral lymph nodes, and, to a<br />
lesser degree, to mesenteric lymph nodes and acute inflammatory sites is impaired.<br />
Mutant mice mounted normal cytotoxic T cell (CTL) responses against systemic LCMV<br />
and VSV infections, and showed normal ex vivo CTL function. However, they did not<br />
reject immunogenic tumors grafted into footpads, and did not demonstrate priming<br />
response against tumor-specific antigen. Thus L deficiency causes a selective defect in<br />
induction of peripheral immune responses, whereas responses to systemic infection are<br />
normal.<br />
M2 and X2: M2 interacts with an assortment of ligands including ICAM-1,<br />
iC3b, fibrinogen, serum factor X, and heparin, and may bind denatured proteins,<br />
deoxyoligonucleotides, elastase, high molecular weight kininogen, and carbohydrate -<br />
glucan structures. It interacts with the 3 rd Ig domain of ICAM-1, whereas L2 interacts<br />
with the 1 st Ig domain. When L2 and M2 are expressed at similar levels, the L2<br />
/ICAM-1 interaction dominates over the M2/ICAM-1 interaction.X2 binds to iC3b,<br />
fibrinogen, and ICAM-1. M2 and X2 mediate myeloid cell adhesion to<br />
endothelium, transmigration, chemotaxis, phagocytosis of opsonized particles, and<br />
respiratory burst. M2-deficient mice have significant reductions in the numbers of<br />
mast cells resident in the peritoneal cavity, peritoneal wall, and dorsal skin. Such mice<br />
exhibit significantly increased mortality to acute septic peritonitis, where host resistance<br />
depends on both mast cells and complement.<br />
D2: D2 binds preferentially to ICAM-3. The D subunit is more closely related to<br />
M and X than to L. D2 is expressed at moderate levels on myelomonocytic cell<br />
lines, and subsets of peripheral blood leukocytes. It is strongly expressed on tissue-
compartmentalized cells such as macrophage foam cells found in aortic fatty streaks that<br />
may develop into atherosclerotic lesions. It is also expressed on eosinophils and binds<br />
VCAM-1, suggesting it may play a role in eosinophil adhesion to VCAM-1 in states of<br />
chronic inflammation. The ICAMs are expressed on dendritic cells, and other antigen<br />
presenting cells, and deliver costimulatory signals via 2 integrins to trigger lymphocyte<br />
proliferation.<br />
E associates with the 7-subunit<br />
The E subunit associates with the 7-subunit, forming the E7 (HML-1, human;<br />
M290, mouse; CD103/7) activation antigen which recognizes epithelial E-cadherin. It<br />
has been proposed to retain iIEL as an immune barrier against intestinal pathogens. E7<br />
is expressed on only 2% of circulating blood lymphocytes and is upregulated by TGF-<br />
a cytokine which may imprint migratory gut-seeking lymphocytes to become resident<br />
as iIELs. The E7 binding site within E-cadherin is distinct from the site used by E-<br />
cadherin for homophilic binding. 7 -/- mice are viable, but have impaired gut-associated<br />
lymphoid tissue including reduced numbers of intraepithelial lymphocytes, and<br />
lymphocytes in the Peyer’s patches, mesenteric lymph nodes, and lamina propria.<br />
Mucosal T lymphocyte numbers are selectively reduced in E -/- mice, including intestinal<br />
and vaginal IEL, and lamina propria lymphocytes. In contrast, peribronchial,<br />
intrapulmonary, Peyer's patch, and splenic T lymphocyte numbers were not reduced,<br />
suggesting that E7 is important for generating or maintaining the gut and vaginal T<br />
lymphocytes located diffusely within the epithelium or lamina propria but not for<br />
generating the gut-associated organized lymphoid tissues.<br />
4 integrins are major immunoreceptors that recognize Ig-family counterreceptors<br />
4 integrins are predominantly expressed on leukocytes, and play key roles in immune<br />
responses. The 4 subunit assembles with the 1 and 7 subunits, producing the integrins<br />
41 (VLA-4) and 47 (LPAM-1), respectively. These two integrins share the ligands<br />
VCAM-1, MAdCAM-1, and FN; where 41 preferentially binds VCAM-1 on activated<br />
endothelium and 47 preferentially binds MAdCAM-1 expressed on high endothelial<br />
venules (HEV) at sites of chronic inflammation. 41 binds to Ig domains 1 and 4 of<br />
VCAM-1, whereas 47 binds to Ig domain 1 of MAdCAM-1 with assistance from<br />
domain 2. Both 4-integrins are expressed on the microvillus tips of lymphocytes, unlike<br />
L2 which is restricted to the planar surface; and can mediate lymphocyte tethering and<br />
rolling under shear flow. 4-integrins also mediate the initial attachment of eosinophils.<br />
41 is more widely expressed, being found outside the leukocyte lineages on myoblasts,<br />
endothelial and melanoma cells. The interaction of 41 with VCAM-1 may facilitate<br />
transendothelial chemotaxis by supporting the lateral migration of attached monocytes<br />
along the endothelium.<br />
Other functions for 41, some of which may be shared with 47, include facilitating<br />
the attachment and emigration of leukocytes across the blood vessel wall, the adhesion<br />
and prevention of apoptosis of B cells in germinal centres, and the adhesion of lymphoid
progenitors to bone marrow stroma. Production of T cells in the adult is 4-dependent,<br />
and precursors for both T and B cells require 4-integrins for normal development within<br />
the bone marrow. Antibodies against 47 or MAdCAM-1 block the binding of<br />
mesenteric lymph node lymphocytes to Peyer’s patch (PP) HEV in vitro, and the homing<br />
of lymphocytes to the gut and lamina propria in vivo. Hence 47 is critical for the<br />
homing of lymphocytes to the gut, and mucosal sites which are normally chronically<br />
inflamed. 4 integrins have an ability to compensate for the lack of LFA-1 in facilitating<br />
the migration of lymphocytes to peripheral lymph nodes in LFA-1-deficient mice.41<br />
also participates in lymphocyte recirculation through bone marrow.<br />
4 integrins, and their ligands are highly expressed on pathogenic leukocytes, and<br />
diseased tissues at sites of chronic inflammation, and hence are major targets for the<br />
treatment of many of the major chronic inflammatory disease. The 4-integrin ligands<br />
VCAM-1 and MAdCAM-1 are expressed on follicular dendritic cells, and may deliver<br />
signals via 4 integrins during antigen presentation, leading to costimulation of T cell<br />
proliferation. <br />
<br />
Ig CAMS<br />
LFA-1<br />
ICAM-3<br />
CD50<br />
Leukocytes activated resting activated HEV<br />
endothelium endothelium endothelium<br />
activated leuk resting lymph/mono macrophage<br />
ICAM-1 (CD54)<br />
Intercellular adhesion molecule-1(ICAM-1) is a single chain 80-114 kDa protein with<br />
five Ig-like domains, a single transmembrane region, and a short cytoplasmic domain. It<br />
can bind to LFA-1, Mac-1, fibrinogen, hyaluronan, and CD43. Resting leukocytes<br />
express little or no detectable ICAM-1, but expression is induced following activation.<br />
Resting endothelial cells have low levels of ICAM-1, and activation with inflammatory
cytokines such as IL-1, IFN-, and TNF-results in increased expression on endothelial<br />
cells in addition to induction on epithelial cells. It is expressed on dendritic cells, where<br />
the interaction of ICAM-1 with LFA-1 is thought to be involved in the initial binding of<br />
T cells to antigen presenting cells.<br />
ICAM-2 (CD102)<br />
ICAM-2 (55-65 kDa) has two Ig-like domains. Resting lymphocytes and monocytes, but<br />
not neutrophils, express low levels of cell surface ICAM-2. Immunohistochemical<br />
analysis indicates high expression of ICAM-2 on endothelium and on follicular dendritic<br />
cells in lymph node germinal centers. ICAM-2 is not upregulated on leukocytes or<br />
cultured endothelium following stimulation with inflammatory mediators. Ligands<br />
recognized by ICAM-2 include LFA-1 and Mac-1. The precise role of ICAM-2 is not<br />
understood, but it is thought to be important in recirculation of leukocytes through<br />
uninflamed tissue endothelium.<br />
ICAM-3 (CD50)<br />
ICAM-3 is a 120-160 kDa molecule and, like ICAM-1, it contains five Ig domains. It is<br />
constitutively expressed at high levels on leukocytes, but, in contrast to ICAM-1 and<br />
ICAM-2, it is not found on most endothelial cells. ICAM-3 binds to LFA-1, but not Mac-<br />
1. It is expressed on dendritic cells, where the interaction of ICAM-3 with LFA-1 is<br />
thought to be involved in the initial binding of T cells to antigen presenting cells.<br />
VCAM-1 (CD106)<br />
Vascular cell adhesion molecule (VCAM-1) (110 kDa) has seven Ig-like domains.<br />
Expression is induced on endothelial cells by inflammatory mediators such as IL-1 and<br />
TNF-. VCAM-1 is also expressed on some macrophages, dendritic cells, bone marrow<br />
stromal cells, synovial cells in inflamed joints, and muscle cells. VCAM-1 is recognized<br />
by the integrins 41 (VLA-4) and 47 and supports the extravasation of leukocytes,<br />
particularly at sites of inflammation. In addition, VCAM-1 can participate in adhesion<br />
outside the vasculature, including binding of lymphocytes to dendritic cells and bone<br />
marrow stromal cells.<br />
PECAM-1 (CD31)<br />
Among the molecules known to (secondarily) activate the integrins are the chemokines<br />
and platelet-endothelial cell adhesion molecule-1 (PECAM-1, CD31 or endoCAM). Once<br />
a leukocyte binds to endothelium via an integrin-immunoglobulin (Ig) superfamily (IgSF)<br />
interaction, it "searches" for junctions between endothelial cells, first squeezing between<br />
these potential discontinuities, and then penetrating the underlying basement membrane<br />
to reach the tissues. Human CD31 is a 130 kDa, type I (extracellular N-terminus)<br />
transmembrane glycoprotein that possesses six Ig-domains. Most, if not all, activities<br />
associated with CD31 can be attributed to homophilic CD31-CD31 interactions. Among<br />
these are leukocyte extravasation, bone marrow hematopoiesis, and vascular<br />
development. During the events surrounding leukocyte extravasation, CD31 seems to be<br />
most important during the later stages. It facilitates the binding of various leukocyteassociated<br />
integrins to ICAM/VCAM IgSF members and assist peripheral blood
leukocytes in their transit across endothelial barriers. On T cells, CD31 ligation will<br />
upregulate the adhesive function of both 1 and 2 integrins.<br />
**************************************************************************************<br />
E-Cadherin<br />
Epithelial cadherin is a 120-kDa cell surface glycoprotein that, when complexed with<br />
catenins, mediates Ca2+-dependent cell-cell adhesion. E-cadherin forms the key<br />
functional component of adherens junctions of all epithelial cells. It plays a major part in<br />
the establishment and maintenance of intercellular adhesion, cell polarity and tissue<br />
architecture. Loss of cadherin mediated adhesion seemed to be an important contributory<br />
factor in tumour pathogenesis. In terms of immunity, it plays a critical role in localizing<br />
intraepithelial lymphocytes to epithelial regions by binding to E7. This is particularly<br />
important for gut mucosal immunity.<br />
**************************************************************************************<br />
Selectins<br />
Selectins are a family of transmembrane molecules, expressed on the surface of<br />
leukocytes and activated endothelial cells. Selectins contain an N-terminal extracellular<br />
domain with structural homology to calcium-dependent lectins, followed by a domain<br />
homologous to epidermal growth factor, and two to nine consensus repeats (CR) similiar<br />
to sequences found in complement regulatory proteins. Each of these adhesion receptors<br />
is inserted via a hydrophobic transmembrane domain and possesses a short cytoplasmic<br />
tail. The initial attachment of leukocytes, during inflammation, from the blood stream is<br />
afforded by the selectin family, and causes a slow downstream movement of leukocytes<br />
along the endothelium via transient, reversible, adhesive interactions called leukocyte<br />
rolling. Each of the three selectins can mediate leukocyte rolling given the appropriate<br />
conditions.<br />
L-selectin<br />
P-selectin<br />
E-selectin
L-Selectin<br />
The smallest of the vascular selectins, a 74-100 kDa molecule, is constitutively expressed<br />
at the tips of microfolds on granulocytes, monocytes, and a vast array of circulating<br />
lymphocytes. L-selectin is important for lymphocyte homing and adhesion to high<br />
endothelial cells of post capillary venules of peripheral lymph nodes. Moreover, this<br />
adhesion molecule contributes greatly to the capture of leukocytes during the early phases<br />
of the adhesion cascade. Following capture, L-selectin is shed from the leukocyte surface<br />
after chemoattractant stimulation. L-selectin interacts with three known counter receptors<br />
or ligands, MAdCAM-1, GlyCAM-1, and CD34.<br />
In L-selectin deficient mice, or following anti-L-selectin mAb treatment, trauma-induced<br />
leukocyte rolling in mesentary or cremaster muscle venules is normal initially, but<br />
declines over time. This indicates that trauma-induced rolling in these mice is P-selectin<br />
dependent with a velocity highly comparable to that observed in wild-type mice. L-<br />
selectin is critical in mediating rolling after surgical trauma and is necessary for<br />
neutrophil recruitment after inflammation. Nevertheless, basal neutrophil trafficking<br />
appears to remain unaffected by absence of L-selectin since peripheral leukocyte and<br />
neutrophil counts in these mice were normal.<br />
Leukocyte rolling is P-selectin dependent in TNF--treated mice deficient in L-selectin,<br />
The lack of L-selectin in these mice reduces the efficiency of E-selectin-mediated rolling<br />
as shown by the sensitivity of rolling to P-selectin antibodies. These experiments<br />
consistently show that L- and P-selectin mediate leukocyte rolling, however, L-selectin<br />
alone cannot assume this task at normal velocities in vivo. L- and P- selectin cooperate in<br />
such a way that in the absence of P-selectin, L-selectin must initiate leukocyte<br />
interactions to allow slow rolling on E-selectin (but see below). In addition, L- or P-<br />
selectin must be present to mediate capture before the commencement of leukocyte<br />
rolling. Without the presence of either L- or P-selectin, rolling cannot occur.<br />
Levels of L-selectin are higher on lymph node-homing T cells, which may facilitate T<br />
cell accumulation at this site by enhancing binding to locally expressed ligands such as<br />
PNAd.<br />
P-selectin<br />
P-selectin is the largest of the known selectins at 140 kDa. It contains nine consensus<br />
repeats (CR), and is expressed in -granules of activated platelets and granules of<br />
endothelial cells. Within minutes of stimulation of the endothelial cells by inflammatory<br />
mediators such as histamine, leukotrienes, thrombin, or phorbol esters, P-selectin is<br />
surface-expressed. The primary ligand for P-selectin is PSGL-1 (P-selectin glycoprotein<br />
ligand-1) which is constitutively found on all leukocytes, and rapidly released from the<br />
cell-surface following leukocyte activation. The transient interactions between P-selectin<br />
and PSGL-1 allow leukocytes to roll along the venular endothelium. Accordingly, P-<br />
selectin is largely responsible for the rolling phase of the leukocyte adhesion cascade. P-<br />
selectin can also mediate capture when L-selectin is not present.
In mice deficient for P-selectin, trauma-induced rolling is absent immediately, but returns<br />
after 1-2 hours. In this case, the delayed rolling is L-selectin dependent, but the<br />
leukocytes roll much faster than in wild-type mice, suggesting that L-selectin cannot<br />
independently support rolling at typical in vivo velocities. On the other hand, in L-<br />
selectin deficient mice, P-selectin mediates most trauma-induced leukocyte rolling.<br />
Normal leukocyte rolling is seen for approximately 90 minutes, showing that P-selectin<br />
has the ability to capture leukocytes from the bloodstream and start them rolling along the<br />
endothelium even without the presence of L-selectin.<br />
In TNF--stimulated venules, P-selectin and E-selectin tend to have overlapping<br />
functions. In mice deficient for P-selectin, it is necessary to block E-selectin function to<br />
significantly reduce rolling, and in E-selectin knockouts, an antibody against P-selectin<br />
must be introduced to reduce rolling. Correspondingly, no leukocyte rolling is observed<br />
in E-selectin/P-selectin double deficient mice treated with TNF-. Although P- and E-<br />
selectin seem to have redundant functions, observations of rolling flux fraction and<br />
rolling velocity indicate that P-selectin is responsible for early rolling while E-selectin<br />
allows slow rolling and more adhesion.<br />
E-selectin<br />
E-selectin is expressed on inflamed endothelial cells in response to treatment with<br />
inflammatory cytokines. Intravital microscopic experiments have shown that its function<br />
in mediating leukocyte rolling is largely redundant with that of P-selectin. Consequently,<br />
E-selectin deficient mice have only a subtle defect in leukocyte rolling as shown by much<br />
faster rolling velocities in these mice. In addition to mediating leukocyte rolling, E-<br />
selectin participates in the conversion of rolling to firm adhesion. E-selectin-deficient<br />
mice have a reduced number of firmly adherent leukocytes in response to local<br />
chemoattractant or cytokine stimulation. This defect may be related to the more rapid<br />
rolling velocities in the absence of E-selectin. E-selectin is expressed in skin microvessels<br />
under baseline conditions, and there is some evidence that E-selectin is of particular<br />
importance in skin inflammation, because it supports the recruitment of skin-specific T<br />
lymphocytes.The ligands for E-selectin that are responsible for the rolling interaction are<br />
unknown. Two candidate ligands, PSGL-1, and E-selectin ligand-1 (ESL-1) have not<br />
been shown to be required for E-selectin mediated leukocyte rolling under any condition.<br />
**************************************************************************************
Mucins<br />
Selectin ligands are mucins. Mucins are serine and threonine-rich, providing sites and a<br />
scaffold for O-link glycosylation.<br />
PSGL-1<br />
P-selectin glycoprotein ligand-1 is a 240 kDa homodimer consisting of two 120kDa<br />
polypeptide chains. PSGL-1 is constitutively expressed on all leukocytes. PSGL-1 is<br />
primarily found on the tips of the microvilli. PSGL-1 can bind to P-selectin on the<br />
endothelium when decorated with appropriate sugars. The structure of functional PSGL-1<br />
includes the sialyl-Lewis x component. The PSGL-1 gene encodes a transmembrane<br />
polypeptide rich in proline, serine and threonine residues typical of mucin-type<br />
glycoproteins. The O-linked glycans displayed by PSGL-1 must undergo two specific<br />
post-translational modifications in order for it to function as a counter-receptor for P-<br />
selectin: (1,3) fucosylation and (2,3) sialylation. PSGL-1 can also bind L-selectin and<br />
E-selectin. CLA the skin homing receptor is a specially modified form of PSGL-1 (see<br />
later).<br />
CD34<br />
CD34 is a transmembrane glycoprotein constitutively expressed on endothelial cells and<br />
on hematopoietic stem cells. This highly O-glycosylated molecule, containing serine and<br />
threonine-rich mucin like domains, binds to L-selectin. Studies have suggested that CD34<br />
is important in tethering lymphocytes. Mice deficient in CD34 exhibited no detectable
abnormalities in postsurgical leukocyte rolling in cremaster venules. Antibodies blocking<br />
L-selectin function reduced rolling in CD34 deficient mice suggesting that CD34 lacks<br />
major significance as a ligand for L-selectin.<br />
**************************************************************************************<br />
Chemoattractants<br />
Extracellular matrix<br />
In a lot of the connective tissues, the extracellular matrix molecules are secreted by cells<br />
called fibroblasts. These molecules assemble into the extracellular matrix once they are<br />
secreted. The extracellular matrix is made up of two classes of macromolecules. The first<br />
class is called glycosaminoglycans, which are polysaccharide chains. Members of this<br />
class are usually found to be covalently linked to protein in the form of proteoglycans.<br />
The second class is made up by fibrous proteins. There are two functional types of<br />
fibrous proteins: the ones that are mainly structural, like collagen and elastin for example,<br />
and the ones that are mainly adhesive, like fibronectin and laminin for example. The<br />
members of the glycosaminoglycans form a highly hydrated, gel-like substance, in which<br />
the members of the fibrous proteins are embedded. Collagen fibers strengthen and help to<br />
organize the matrix, while elastin fibres give it resiliance. The adhesive proteins help<br />
cells to attach to the extracellular matrix. Fibronectin for example promotes the<br />
attachment of fibroblasts and other cells to the matrix in connective tissues via the<br />
extracellular parts of some members of the integrin family, while laminin promotes the<br />
attachment of epithelial cells to the basal lamina, again via the extracellular domains of<br />
some members of integrins.<br />
**************************************************************************************<br />
Multistep process of leukocyte extravasation<br />
The stimulus for endothelial activation in vivo is probably local production of cytokines<br />
and other inflammatory mediators released on tissue injury.<br />
Capture<br />
The process known as capture or tethering represents the first contact of a leukocyte with<br />
the activated endothelium. Capture occurs after margination, which allows leukocytes to<br />
move in a position close to the endothelium, away from the central blood stream. During<br />
the inflammatory response, endothelial activation is required to initiate capture.<br />
P-selectin on endothelial cells, is the primary adhesion molecule for capture and the<br />
initiation of rolling. The main leukocyte ligand for P-selectin is PSGL-1. In addition,<br />
many in vivo studies suggest that L-selectin exhibits an important role in capture as well.<br />
Antibodies blocking L-selectin function inhibit rolling in many models in which rolling is<br />
P-selectin dependent.<br />
EFigure 10. Expression of ICAM-3 (CD50)<br />
E-cadherin.10)
Rolling<br />
Once leukocytes are captured, they may transiently adhere to the venular endothelium<br />
and begin to roll. Rolling occurs at or below the velocity of freely flowing cells. The<br />
velocity separating rolling from freely flowing cells is called critical velocity or<br />
hydrodynamic velocity. P-selectin is the most important selectin involved in rolling. P-<br />
selectin can support both capture and rolling in the absence of L-selectin. Although P-<br />
selectin was initially identified on activated platelets, it is also found in Weibel-<br />
Palade<br />
The multistep process of leukocyte extravasation<br />
Margination<br />
Luster et al. Nature Immunology 6: 1182-1190, 2005<br />
bodies of human endothelial cells. Upon stimulation by trauma, P-selectin is rapidly<br />
surface-expressed on the venular endothelium, and it makes the endothelium "sticky" to<br />
leukocytes. PSGL-1 (P-Selectin Glycoprotein Ligand-1) is constitutively expressed on all<br />
lymphocytes, monocytes, eosinophils, and neutrophils. PSGL-1 on neutrophils,<br />
eosinophils, and monocytes has a glycosylation pattern allowing it to bind to endothelial<br />
P-selectin. As a result, the leukocyte rolls along the endothelium. During rolling, bonds<br />
are formed at the leading edge of the rolling cell and broken at the trailing edge.<br />
Leukocyte integrins initially remain in their resting state, and endothelial IgCAMs remain<br />
at control levels.<br />
The critical role of P-selectin in the rolling phase of the adhesion cascade is supported in<br />
experiments done on gene-targeted mice. In mice lacking P-selectin, leukocytes do not<br />
roll on the venular endothelium after surgical trauma. Also, when compared with wildtype<br />
mice, the number of circulating granulocytes is greater in P-selectin deficient mice.<br />
This suggests that P-selectin is necessary to remove the leukocytes from the bloodstream
so they may stick and roll along the venular endothelium. In vitro, isolated human<br />
granulocytes have been shown to roll on purified P-selectin using flow chamber systems.<br />
L-selectin and E-selectin also take part in the rolling process. When P-selectin is absent,<br />
trauma-induced rolling becomes L-selectin dependent, but the average leukocyte rolling<br />
velocity is three to five times faster in this case. This suggests that L-selectin is much less<br />
efficient than P-selectin in mediating the rolling process. However, L-selectin is<br />
necessary for the normal inflammatory response in capturing leukocytes and initiating<br />
rolling. An apparent redundancy exists between P- and E-selectin in mediating leukocyte<br />
rolling on cytokine-activated endothelium. E-selectin is thought to be responsible for<br />
slow rolling interactions, and possibly the initiation of firm adhesion.<br />
Slow rolling<br />
After induction of inflammation by injection of a pro-inflammatory cytokine like TNF-,<br />
leukocyte rolling velocity drops dramatically to an average between 5 and 10 µm/s. This<br />
rolling requires the expression of E-selectin on endothelial cells and CD18 integrins on<br />
the rolling leukocytes, and has been termed "slow rolling" to distinguish it from the much<br />
faster rolling without cytokine stimulation. Slow rolling is not based on a unique property<br />
of E-selectin, but the expression of E-selectin and/or its ligands appears to be sufficiently<br />
high in vivo to support slow rolling. The contact time during which the leukocyte is in<br />
close proximity with the endothelium, appears to be a key parameter in determining the<br />
success of the recruitment process as reflected in firm adhesion. The important role of<br />
leukocyte transit time appears to be related to chemokines that are presented on the<br />
endothelial surface and are likely to be accessible to the leukocyte as long as it rolls. This<br />
is supported by tracking studies, in which individual rolling leukocytes were found to<br />
slow down systematically before becoming firmly adherent. Rolling leukocytes are likely<br />
to be activated by surface-bound chemoattractants and through adhesion molecule-based<br />
signaling. It is also possible that the velocity of rolling may have an effect independent of<br />
transit time, because secondary binding events (e.g., 2 integrin-mediated) may not be<br />
able to form unless the leukocyte spends a certain amount of time in a position favorable<br />
for bond formation. Although slow rolling makes leukocyte recruitment much more<br />
efficient, it is not strictly required, because high concentrations of chemoattractants can<br />
also arrest fast-rolling leukocytes. Chemokines activate integrins resulting in firm<br />
adhesion of leukocytes to the vascular wall.<br />
Firm adhesion<br />
It is thought that most if not all leukocytes adhere only after having rolled. Several<br />
studies suggest that direct adhesion (from the free-flowing leukocyte pool) is extremely<br />
rare. E-selectin participates in the conversion of rolling to firm adhesion. E-selectin<br />
deficient mice have a reduced number of firmly adherent leukocytes in response to local<br />
chemoattractant or cytokine stimulation.<br />
Interfering with CD18 integrin function is one of the most efficient ways to curb<br />
leukocyte recruitment in many forms of experimental inflammation. Although the<br />
response to exogenous chemoattractant is drastically reduced when CD18 is absent or not<br />
functional, cytokine treatment still yields a robust inflammatory response in gene-
targeted mice lacking CD18. This suggests that CD18 integrins participate in leukocyte<br />
arrest, but are not always required. Neutrophils express small amounts of other integrins,<br />
including 41 integrin, which may be important in these alternative pathways.<br />
However, CD18 deficient mice have severe inflammatory defects including skin<br />
ulcerations, elevated neutrophil counts and immunoglobulin levels, increased<br />
susceptibility to streptococcus pneumoniae, and a severe defect in leukocyte adhesion and<br />
T-cell activation, a defect in leukocyte recruitment to peritonitis, and a lack of neutrophil<br />
recruitment to the skin. Patients lacking CD18 expression suffer from leukocyte adhesion<br />
deficiency type 1 (LAD-1). When CD18 is totally absent, LAD-1 is a very severe disease,<br />
which can lead to early lethality.<br />
Leukocytes rolling in resting inflamed venules require ICAM-1 to stop in response to a<br />
chemoattractant. However, ICAM-1 is no longer required after activation with<br />
inflammatory cytokines. Monocytes, eosinophils, and many lymphocytes express 41<br />
integrin (VLA-4), and mouse and rat neutrophils also express small amounts. When other<br />
adhesion molecules are unavailable, 41 integrin can mediate both leukocyte rolling,<br />
and firm adhesion. 41 integrin binds to endothelial VCAM-1, and alternatively spliced<br />
fibronectin. Intravital microscopic evidence available to-date suggests that most 4-<br />
dependent binding is through VCAM-1, because antibodies to VCAM-1 applied in the<br />
microcirculation and in atherosclerotic arteries block leukocyte rolling and adhesion to a<br />
similar extent as 4 antibodies.<br />
Transmigration<br />
Leukocytes migrate across resting endothelium if an exogenous chemoattractant is<br />
present. This has been termed "leukocyte driven" or chemotactic transmigration. The<br />
pathophysiological hallmark of established inflammatory transmigration is endothelial<br />
activation, an event requiring transcription and protein synthesis. As a result adhesion<br />
molecules are upregulated, inflammatory mediators are produced, and the endothelium<br />
secretes chemoattractants, all of which contribute to transmigration. Endothelial<br />
chemokines are critical for transmigration. Chemokines have heparin binding sites which<br />
allow them to bind proteoglycans on the vascular endothelium, positioning them for<br />
activating integrins. A number of adhesion molecules have been implicated in<br />
transmigration, although the level of confidence in their actual involvement varies,<br />
including PECAM-1, ICAM-1, VE-cadherin, CD11a/CD18 (LFA-1), IAP (CD47) and<br />
VLA-4 (41 integrin). The cells upon activation by integrins release heparin binding<br />
protein (HBP) which causes increases in microvascular permeability which allows for<br />
leukocyte transmigration.<br />
In vivo, ICAM-1 knock-out mice show significantly impaired neutrophil migration into<br />
inflamed peritoneum, and ICAM-1 antibodies reduce acute and chronic inflammation in a<br />
number of animal models. ICAM-1 antibodies reduce cytokine-activated transmigration<br />
of neutrophils in vitro, by over 85%. ICAM-1 is also important in chemotactic<br />
transmigration. Antibodies inhibit neutrophil chemotactic transmigration by ~55%.<br />
PECAM-1 (Platelet-Endothelial Cell Adhesion Molecule-1, CD31) is critical in passage<br />
through the junction in cytokine-activated transmigration. Anti-PECAM-1 antibodies<br />
reduce monocyte transmigration through resting endothelium, and both monocyte and
neutrophil transmigration through cytokine activated endothelium by 70-90%. Binding of<br />
leukocytes to endothelium is not affected. Although V3 integrin can be a ligand for<br />
PECAM-1, and monocytes lacking 3-integrin transmigrate poorly, this appears to be<br />
due to modulation of CD11a/CD18 rather than by an interaction with PECAM-1. In<br />
contrast to cytokine-activated transmigration, PECAM-1 seems to have little role in<br />
chemotactic transmigration. PECAM-1 antibodies do not decrease chemotactic<br />
transmigration in vivo, nor do they decrease transmigration triggered by thrombin. In<br />
addition, there is often a significant residual transmigration (~10-30%) after PECAM-1<br />
inhibition which suggests that other mechanisms may operate in passage through the<br />
junction in cytokine-activated transmigration. These observations suggest that the<br />
mechanisms operating in cytokine-activated and chemotactic transmigration overlap to<br />
only a small degree. Neutrophils and monocytes from CD11a knock out mice do not<br />
require PECAM-1 for transmigration, suggesting that CD11a and PECAM-1 somehow<br />
operate in a similar pathway of migration. However, PECAM-1 knockout mice show no<br />
clear evidence of a transmigration defect.<br />
The involvement of VLA-4 in transmigration in vitro is variable. It is likely this reflects<br />
redundancy with 2-integrins, as well as the degree of VCAM-1 expression on<br />
endothelium, which in turn reflects the state of endothelial activation. It appears<br />
unimportant in neutrophil transmigration in vitro, and in transmigration of activated T-<br />
cells. Resting T-cell and NK cell cytokine-activated transmigration are reduced by 40%<br />
and 30% respectively (Oppenheimer Marks et al 1991; Bianchi et al 1993). As firm<br />
adhesion is reduced to a similar extent it is unclear whether this is a specific effect on<br />
passage across the endothelium.<br />
**************************************************************************************<br />
Ectoenzymes in the control of leukocyte traffic<br />
Ectoenzymes are membrane proteins that have their active site outside the cell. They<br />
include proteases, nucleotidases, and oxidases, which can regulate leukocyte<br />
transmigration.<br />
Nucleotidases:<br />
ATP is proinfammatory: ATP binds purinergic receptors of the P2X and P2Y<br />
families, and induces proliferation by inducing cytokine expression, and activating<br />
dendritic cells.<br />
Adenosine is anti-infammatory: Adenosine inhibits neutrophil adhesion to the<br />
microvascular endothelium by activating the adenosine receptors A 2A R abd A 2B R on<br />
neutrophils. Adenosine prevents leukocyte activation- it inhibits L-selectin shedding and<br />
expression of CD18 integrins by leukocytes, and down-regulates VCAM-1 and cytokine<br />
expression by endothelial cells.<br />
CD73
CD73 is a glycosylphosphatidylinositol (GPI)-linked cell surface molecule expressed by<br />
vascular endothelial cells and up to 15% of lymphocytes (but not by granulocytes and<br />
monocytes). It catalyses the dephosphorylation of AMP to adenosine, indicating it has<br />
anti-inflammatory properties. CD73 is increased in expression at sites of inflammation by<br />
IFN. Mice deficient in CD73 show increased leukocyte attachment to the endothelium,<br />
and leukocyte migration.<br />
CD39<br />
CD39 converts extracellular ATP to AMP, suggesting it has anti-inflammatory<br />
properties. It is expressed by many different types of leukocytes, and by vascular<br />
endothelial cells. Mice deficient in CD39 display exacerbated skin inflammation by<br />
irritants.<br />
Both ATP-generating and ATP-consuming pathways coexist on the surfaces of<br />
leukocytes and endothelial cells. Their balance regulates the level of ATP and adenosine,<br />
and hence influence the local inflammatory environment.<br />
ATP is dephosphorylated to AMP by CD39 on resting endothelium, and AMP is<br />
dephosphorylated to adenosine by CD73. This is expected to create an anti-inflammatory<br />
setting. The activity of CD73 is inhibited when lymphocytes attach to the endothelium,<br />
and hence less adenosine is produced, which leads to increased leukocyte migration.<br />
ADP-ribosyltransferases:<br />
ADP-ribosyltransferases such as ART2 transfer the ADP-ribose moiety from NAD(P) to<br />
an acceptor. The ADP-ribose moiety can covalently modify and inactivate several surface<br />
proteins including L2. The homing of NAD(P)-treated T cells to lymph nodes and gut
lymphoid organs is inhibited, and injection of NAD(P) into mice markedly decreases T<br />
cell homing. Note: 41 is not ADP-ribosylated, and B cells don’t express ART2, hence<br />
their homing is not affected.<br />
Ectopeptidases regulate chemokines<br />
CD26 is an ectoenzyme that cleaves N-terminal dipeptides containing either proline or<br />
alanine residues from polypeptides. CD26 is expressed de novo on activated T and B<br />
cells, NK cells, and endothelial cells. Numerous bioactive peptides are cleaved by CD26,<br />
including chemokines. Cleavage of chemokines by CD26 reduces their ability to bind and<br />
activate chemokine receptors, whereas their ability to serve as chemoattracts can be<br />
enhanced (eg CCL5). Rodents that are deficient in CD26 have increased infiltration of<br />
leukocytes into the lung and joints in models of asthma and arthritis, suggesting that<br />
CD26 plays a role in inhibiting inflammation.<br />
Sheddases<br />
CD156b is a sheddase that cleaves L-selectin from the surface of thymocytes. Other<br />
sheddases are also involved. L-selectin is shed from lymphocytes to prevent re-entry of<br />
activated T cells to secondary lymphoid organs.<br />
Ectoenzymes in the control of leukocyte traffic<br />
The nucleotidases CD39 and CD73 regulate the balance of ATP and adenosine, and the<br />
activation of leukocyte integrins and vascular cell adhesion molecules. CD26<br />
proteolytically modifies the activities of chemokines, whereas sheddases cleave leukocyte<br />
cell adhesion molecules such as L-selectin. ADP ribosyltransferases such as ART2<br />
modify and inactivate adhesion molecules.
Poorly characterized<br />
**************************************************************************************<br />
Trafficking of leukocytes to specific organs and tissues, and<br />
microenvironment-specific homing<br />
Tissue-restricted recirculation of memory and effector lymphocytes may serve to 1)<br />
increase the efficiency of regional immune responses, and 2) allow functional immune<br />
specialization of particular tissues. Differences in chemokine receptor, integrin, or<br />
selectin expression plays a major role in selective leukocyte recruitment to different<br />
tissues or organs, and positional localization with an organ. Chemokines play a major<br />
role as they serve as attractants for inflammatory leukocytes, control leukocyte/integrin<br />
activation, and control microenvironmental segregation within lymphoid organs. Table 1<br />
shows examples of key molecules that control recruitment. Different chemokine<br />
receptors are expressed depending on whether the T cell is naïve, or a memory cell, and<br />
whether it is a Th1 or Th2 cell.<br />
Table 1<br />
Receptors on leukocytes that contribute to selective cell<br />
recruitment<br />
Leukocyte surface structure Ligands Pattern of leukocyte expression<br />
Adhesion molecules
L-selectin PNAd, others All leukocytes, but naive T cells > memory T cells<br />
PSGL-1 P-selectin All leukocytes, but eosinophils > neutrophils<br />
CLA E-selectin Skin-homing memory T cells<br />
Sialyl-dimeric Lex E-selectin All leukocytes, but neutrophils > eosinophils<br />
41 integrin VCAM-1, others All leukocytes except neutrophils<br />
47 integrin MAdCAM-1, others All leukocytes except neutrophils<br />
E7 integrin E-cadherin Intraepithelial lymphocytes<br />
Chemokine receptors<br />
CCR3 Eotaxin 1-3, others Eosinophils, basophils, mast cells; some TH2 cells<br />
CCR4 TARC, MDC Skin-homing memory T cells<br />
CCR5 MIP-1 TH1 cells<br />
CCR6 MIP-3 Memory T cells<br />
CCR7 SLC (CCL21), Naive B and T cells<br />
MIP-3 (CCL19)<br />
CCR8 I-309 TH2 cells<br />
CCR9 TECK Gut-homing memory T cells<br />
CCR10 CTACK Skin-homing memory T cells<br />
CXCR3 Mig, IP-10, I-TAC TH1 cells<br />
CXCR5 BLC (CXCL13) Naive B cells<br />
PNAd, Peripheral lymph node addressin; PSGL-1, P-selectin glycoprotein ligand-1; CLA, cutaneous<br />
lymphocyte antigen; Lex, Lewis X; VCAM-1, vascular cell adhesion molecule-1; MAdCAM-1, mucosal<br />
addressin adhesion molecule-1; TARC, thymus- and activation-related chemokine; MDC, macrophagederived<br />
chemokine; MIP, macrophage inflammatory protein; SLC, secondary lymphoid tissue chemokine;<br />
TECK, thymus-expressed chemokine.
Guidance of neutrophils to sites of sterile inflammation<br />
A multistep cascade of molecular events guides the recruitment of neutrophils to sites of<br />
sterile injury ie tissue injury in the absence of infection. Neutrophils contribute to the<br />
clearance of debris from necrotic sites. They have an arsenal of destructive molecules<br />
hence collateral tissue destruction needs to be avoided. Steps:<br />
1. ATP released from necrotic cells at the wound site activates the NLRP3<br />
inflammasome via the P2X7 receptor in inflammatory cells (macrophages)<br />
resulting in release of IL-1.<br />
2. IL-1 upregulates ICAM-1 on the vascular endothelium, leading to M2<br />
integrin-mediated adhesion of neutrophils to microvascular endothelia near the<br />
wound site within 30-60 minutes of injury.<br />
3. The chemokine MIP-2 (CXCL2) released at the wound site forms a<br />
chemoattractant gradient that surrounds but not does completely reach the injured<br />
area. It is maximal at 100-150 m from the injury border.<br />
4. Neutrophils migrate via an intravascular route to the site of injury.<br />
5. MIP-2 attacts neutrophils up to the injury border (but what attracts neutrophils<br />
into the necrotic lesion?).<br />
6. Necrotic cells release mitochondria-derived formylated peptides, which over-ride<br />
MIP-2 and guide neutrophils directly into the injury site via formyl peptide<br />
receptor 1 signalling. This focuses the innate immune response on sites of damage<br />
rather than healthy tissue.<br />
Did this pathway evolve to limit collateral damage by allowing neutrophils to remain<br />
intravascular during migration to the wound via healthy tissue? The pathway has been<br />
established in liver, but also applies to skin.<br />
Figure MIP-2 expression (red)<br />
and PECAM-1+ endothelium<br />
(blue) were visualized 2.5 hours<br />
after injury.
Homing of lymphocytes to lymph nodes<br />
Naïve T cells: Naïve T cells interact with high endothelial venules in lymph nodes via<br />
L-selectin binding to PNAd, and LFA-1 binding to ICAM-1. These interactions are<br />
stimulated by the chemokine SLC (displayed by HEV), which interacts with CCR7 on the<br />
T cell. The positional localization of naïve T cells within the lymph node is controlled by<br />
the production of MIP-3 within the T cell zone. Thus, the development of lymph nodes<br />
is markedly reduced in L-selectin-deficient mice, such that there are few B and T cells.<br />
The plt (“paucity of lymph node T cells”) mutant mouse deficient in SLC has lymph<br />
nodes and Peyer’s patches that are deficient in T cells, but not B cells. B cells are not<br />
dependent on SLC for entry through HEV. Normal T cells infused into these mice fail to<br />
accumulate in lymph nodes. Injection of SLC into mice leads to uptake and display of<br />
SLC on HEV of lymph nodes, restoring T cell homing. SLC is only prominently<br />
expressed in the T cell zone of lymph nodes. SLC triggers integrin-dependent arrest of<br />
CCR7-bearing T cells. Th1 and Th2 cells may home to different regions within the lymph<br />
node, where CCR7 is more consistently expressed by Th1 cells. MIP-3 is not found on<br />
HEV, but is present in the T cell area. It is thought to play a role in positional<br />
localization rather than capture. Memory effector T cells lack CCR7, and can’t enter<br />
lymph nodes. They home to inflammatory sites in non-lymphoid organs where they play<br />
a role in antigen-specific immune responses.<br />
Naïve B cells: CXCR5, a ligand for the chemokine BLC is found predominantly on<br />
naïve B cells. BLC has been localized to the B cell follicles of lymph nodes, spleen, and
Peyer’s patches. Mice deficient in CXCR5 have diminished development of B cell<br />
follicles. Ectopic expression of BLC in pancreatic islets in mice, results in local B cell<br />
accumulation. Granulocytes don’t home to lymph nodes, presumably because they don’t<br />
express CCR7 and CXCR5.<br />
Chemokines control microenvironmental segregation within lymphoid<br />
organs<br />
Thus, the activation of B and T cells in lymph node HEV is different, and may initiate the<br />
segregation into B and T cell zones. CXCR5 or CCR7-deficient animals have<br />
disorganized lymphoid organs. This indicates that chemokines are key factors in the<br />
development and maintenance of segregated microenvironments characteristic of<br />
secondary lymphoid organs, and in the movement of lymphocytes in and out of these<br />
areas. CCR7 and CXCR5 may be involved in the organization of T and B cell zones.<br />
Balanced reponsiveness to chemokines from adjacent zones determines B<br />
cell movement to the T cell zone<br />
After B cells bind antigen they move to the boundary of B and T cell zones to interact<br />
with T-helper cells. Antigen-engaged B cells have increased expression of CCR7, and<br />
exhibit increased responsiveness to the T cell zone chemokines MIP3, and SLC. In mice<br />
lacking MIP3 and SLC, or CCR7, B cells fail to move to the T cell zone after antigen<br />
engagement. Increased expression of CCR7 by retroviral transfer is sufficient to direct B<br />
cells to the T cell zone. Normally, antigen-stimulated B cells respond less strongly than T<br />
cells to MIP3, and SLC, and are excluded from the central T cell zone. Conversely<br />
overexpression of CXCR5 is sufficient to overcome antigen-induced B cell movement to<br />
the T cell zone. Thus, lymphocyte localization can be determined by the balance of<br />
responsiveness to chemoattractants made in separate but adjacent zones.<br />
From Reif et al. Nature 416: 94-99,<br />
2002<br />
CCL19 = MIP3<br />
CCL21 = SLC<br />
CXCL13 = BLC<br />
Homing of memory T cells and eosinophils to skin<br />
Memory T cells, which express CD45RO, can enter any inflamed tissue to perform<br />
antigen-specific immunosurveillance. They are selectively recruited after allergen<br />
challenge of the skin. Eosinophils also accumulate in the skin.<br />
Memory lymphocytes: CLA (specialized form of PSGL-1), which is a ligand for E-<br />
selectin, is only found on activated memory skin homing T cells. E-selectin is induced de<br />
novo on inflamed postcapillary endothelium, especially in skin. Recruitment of memory<br />
CLA + T cells to the skin during allergic cutaneous inflammation involves interactions by
CLA binding E-selectin, LFA-1 to ICAM-1, VLA-4 to VCAM-1, which are stimulated<br />
by endothelial-displayed TARC binding to CCR4 on leukocytes. Subsequent localization<br />
within tissues may then be controlled by local production of other chemokines such as<br />
MDC binding to CCR4, and CTACK binding to CCR10, and by VLA-1 and VLA-2.<br />
Humans suffering from leukocyte adhesion deficiency syndrome-1 (LAD-1), due loss of<br />
2 subunit, have frequent skin infections. The skin is devoid of neutrophils, and there are<br />
reduced numbers of other leukocytes. CCR4 is highly expressed on CLA + memory T<br />
cells. Its ligand TARC is present in venules of inflamed skin. TARC is expressed by<br />
endothelial cells and stimulates T cell adhesion to ICAM-1. CTACK preferentially<br />
activates homing to the skin of memory CLA + T cells. It is produced by keratinocytes<br />
within the skin, and activates cells via CCR10. CCR4 and CCR10 may have overlapping<br />
roles in cutaneous lymphocyte recruitment. CTACK may be transcytosed, and presented<br />
on endothelium to support triggering of adhesion of skin-homing lymphocytes.<br />
Eosinophils: Eosinophil migration into the skin during allergic cutaneous inflammation<br />
involves P-selectin binding to PSGL-1, LFA-1 binding to ICAM-1, and VLA-4 binding<br />
to VCAM-1, probably stimulated by eotaxin and eotaxin-3 binding to CCR3. Subsequent<br />
localization may be mediated by CCR3-active chemokines, and by VLA-4, and VLA-6.<br />
Human eosinophils express high levels of PSGL-1 and L-selectin. CCR3-active<br />
chemokines such as RANTES, eotaxin, eotaxin-2, and MCP-4 are implicated in<br />
eosinophil accumulation in skin. Intradermal injection of RANTES, or eotaxin-3 causes<br />
selective and rapid accumulation of eosinophils.
Sunshine, vitamin D3, and dendritic cells imprint T cells to migrate to the<br />
epidermis of the skin<br />
The upper layer of the skin generates vitamin D3 from 7-dehydrocholesterol in response<br />
to UVB rays. Dendritic cells in the skin express the vitamin D3 hydroxylases CYP27A1<br />
and CYP27B1 which metabolize vitamin D3 to its active form 1,25(OH)2D3.<br />
1,25(OH)2D3 stimulates memory or effector T cells to express the chemokine receptor<br />
CCR10. CCR10-expressing T cells are attracted to the epidermis by the epidermal<br />
chemokine CCL27 (CTACK), which is expressed by keratinocytes. It is thought this<br />
system evolved to cope with frequent sun exposure and UV damage to the outer layers of<br />
the skin.<br />
Note: CCR10 is not required to recruit T cells to the dermis as this can be achieved by<br />
CCR4. Rather CCR10 is required to attract T cells from the dermis to the epidermis.<br />
Skin DCs migrate via the lymphatics to peripheral lymph nodes where they might imprint<br />
naïve T cells with a skin homing phenotype (CLA + CCR4 hi 47 - ).<br />
1,25(OH)2D3 inhibits the spontaneous upregulation of 47 on activated T cells, and<br />
inhibits the induction of 47 and CCR9 by retinoic acid.<br />
Leukocyte homing to the gut<br />
Gut associated lymphoid tissues include Peyer’s patches (secondary lymphoid organs<br />
resembling lymph nodes), mesenteric lymph nodes, and the lamina propria. Homing of<br />
lymphocytes to Peyer’s patches and lamina propria involve interactions between LFA-1<br />
and ICAM-1, 47 and MAdCAM-1, MAdCAM-1 and L-selectin, probably stimulated<br />
by TECK binding to CCR9 or SLC binding to CCR7. Chemokines such as IP-10,<br />
RANTES, MIP-1, and MIP-1 may influence lymphocyte accumulation within the<br />
lamina propria under both inflamed and uninflamed conditions. A subset of gut-homing T
cells express CCR5 and CXCR3 and E7 and can migrate to the gut epithelium where<br />
they interact with E-cadherin. For B cells, local production in Peyer’s patches of BLC<br />
may be important. Accumulation of eosinophils involves LFA-1/ICAM-1, VLA-<br />
4/VCAM-1, and 47/MAdCAM-1/L-selectin, perhaps influenced by eotaxin produced<br />
by mononuclear cells.<br />
Animals deficient in 7 integrins fail to accumulate T cells in the gastrointestinal tract,<br />
and Peyer’s patches, because 47 is missing. SLC (which binds CCR7) stimulates<br />
47-mediated adhesion of lymphocytes to MAdCAM-1. Defects in CCR7 or SLC<br />
produce mice with reduced numbers of T cells in secondary lymphoid organs, including<br />
Peyer’s patches. For B cells, BLC is important, as mice deficient in the BLC receptor<br />
CXCR5 fail to develop normal Peyer’s patches. But SLC and BLC are not restricted to<br />
the gut, hence are important in homing to lymph nodes in general. TECK, however, is<br />
produced by small intestinal epithelial cells, but not by skin or lymph nodes, or by other<br />
regions of the colon. It is mostly expressed in the crypt region most closely associated<br />
with MAdCAM-1 expressing vessels. TECK has been detected on small intestinal<br />
endothelium. Its receptor CCR9 is expressed on virtually all T cells in the small intestine,<br />
and on memory CD4+ T cells, especially those expressing high levels of 47, whereas<br />
it is not on other memory CD4 + T cells. The GI tract represents the largest reservoir of<br />
eosinophils within the body. Eosinophils have the same pattern of adhesion molecules,<br />
but don’t express either E7, or the chemokine receptors found on gut-homing T cells.<br />
CCR3-active chemokines appear to be responsible for eosinophil accumulation. Eotaxindeficient<br />
mice have few eosinophils within the villae of the intestine.
Vitamin A and dendritic cells imprint T and B cells to migrate to the small<br />
intestine<br />
The small intestine absorbs vitamin A as retinol and processes it to retinoic acid which is<br />
present at high concentrations in the gut wall. Lumenal antigens in the gut are<br />
internalized by M cells, and processed by DCs in the lamina propria. DCs in the gut are<br />
induced by gut TGF- to express E7, and they express enzymes capable of processing<br />
retinol to retinoic acid. E7+ DCs activate naive T and B cells in the mesenteric lymph<br />
nodes (MLN) to express 47 and CCR9 in a retinoic acid-dependent fashion, thereby<br />
imprinting the T and B cells with small intestine-homing properties. Retinoic acid<br />
inhibits the induction of the skin homing receptor CLA.<br />
Acknowledgement: Partly taken from http://hsc.virginia.edu/medicine/basicsci/biomed/ley/index.html,<br />
R&D Systems; Springer TA Annu Rev Physiol 57: 827-872,<br />
1995; Bochner BS J Allergy Clin Immunol 106: 817-828, 2000; Salmi M and Jalkanen S<br />
Nature Rev Immunol 5: 760771, 2005; and Sigmundsdottir H, Butcher EC. Nat Immunol.<br />
9: 981-7, 2008.<br />
Table 1. Leukocyte integrins and their ligands
L2 Leukocytes ICAM-1, ICAM-2, ICAM-3<br />
Telencephalin, Type I collagen<br />
Landsteiner-Wiener (LW) blood<br />
group glycoprotein<br />
M2 Leukocytes ICAM-1, iC3b, factor X, galectin-3, Cfibrinogen<br />
complement factor H, CD23,<br />
neutrophil inhibitory factor,<br />
oligodeoxynucleotides,<br />
heparin, elastase, -glucans,<br />
high molecular weight kininogen,<br />
myeloperoxidase, azurocidin,<br />
haptoglobin, denatured proteins,<br />
Landsteiner-Wiener (LW) blood<br />
group glycoprotein, fibrinogen,<br />
Lipopolysaccarides, pertussis toxin<br />
X2 Leukocytes iC3b, fibrinogen, CD23,<br />
lipopolysaccarides<br />
D2 Macrophages, eosinophils ICAM-3, VCAM-1<br />
<br />
41 Leukocyte, muscle, VCAM-1, FN, MAdCAM-1<br />
stimulated neutrophils,<br />
neural crest cells,<br />
fibroblasts<br />
TSP, osteopontin, chondroitin<br />
sulfate glycosaminoglycan<br />
Propolypeptide of von Willebrand factor<br />
casein, denatured albumin, 4 subunit<br />
47 Leukocytes MAdCAM-1,VCAM-1, FN<br />
E7 Activated leukocytes, IEL E-cadherin, RGD proteins?