

Semi-Batch Emulsion Copolymerization of Vinyl Acetate and Butyl ...
Semi-Batch Emulsion Copolymerization of Vinyl Acetate and Butyl ...
Semi-Batch Emulsion Copolymerization of Vinyl Acetate and Butyl ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
2614<br />
Macromol. Chem. Phys. 2001, 202, 2614–2622<br />
Full Paper: Detailed kinetic studies on semi-batch emulsion<br />
copolymerization <strong>of</strong> vinyl acetate/butyl acrylate<br />
(VAc/BuA) (80:20) at 808C were carried out using alkyl<br />
polyglucoside nonionic surfactants <strong>and</strong> ammonium persulfate<br />
as initiator. The polymerization recipe was varied<br />
with respect to the amount <strong>of</strong> initiator initially charged<br />
<strong>and</strong> continuously fed to the reactor, the surfactant concentration,<br />
the electrolyte concentration, <strong>and</strong> the monomer<br />
addition rate. Various types <strong>of</strong> alkyl polyglucosides with<br />
different hydrophilic <strong>and</strong> hydrophobic chain lengths were<br />
tested. Latex particle stabilization was shown to depend<br />
significantly on the surfactant structure <strong>and</strong> its adsorption<br />
characteristics. In general, the latex particle size distribution<br />
evolved from a bimodal to a unimodal distribution<br />
while latex destabilization was observed at high surfactant<br />
or/<strong>and</strong> high initiator concentrations. Based on experimental<br />
observations it was found that the most important factor<br />
for imparting particle stability was the hydrophilic/<br />
hydrophobic chain length ratio <strong>of</strong> the surfactant which<br />
actually controlled the balance between surface coverage<br />
<strong>and</strong> stabilizing capability.<br />
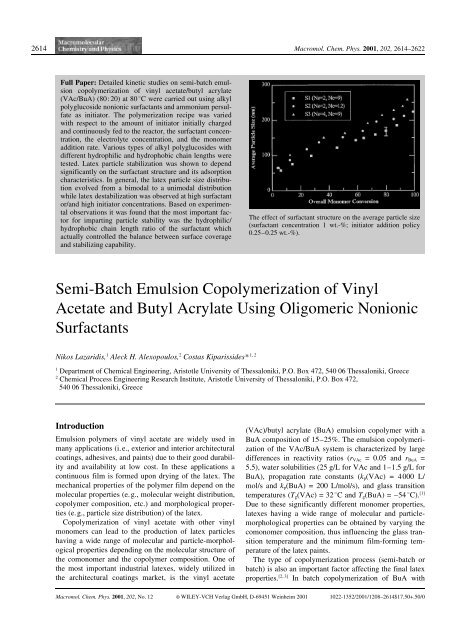
The effect <strong>of</strong> surfactant structure on the average particle size<br />
(surfactant concentration 1 wt.-%; initiator addition policy<br />
0.25–0.25 wt.-%).<br />
<strong>Semi</strong>-<strong>Batch</strong> <strong>Emulsion</strong> <strong>Copolymerization</strong> <strong>of</strong> <strong>Vinyl</strong><br />
<strong>Acetate</strong> <strong>and</strong> <strong>Butyl</strong> Acrylate Using Oligomeric Nonionic<br />
Surfactants<br />
Nikos Lazaridis, 1 Aleck H. Alexopoulos, 2 Costas Kiparissides* 1, 2<br />
1<br />
Department <strong>of</strong> Chemical Engineering, Aristotle University <strong>of</strong> Thessaloniki, P.O. Box 472, 540 06 Thessaloniki, Greece<br />
2<br />
Chemical Process Engineering Research Institute, Aristotle University <strong>of</strong> Thessaloniki, P.O. Box 472,<br />
540 06 Thessaloniki, Greece<br />
Introduction<br />
<strong>Emulsion</strong> polymers <strong>of</strong> vinyl acetate are widely used in<br />
many applications (i.e., exterior <strong>and</strong> interior architectural<br />
coatings, adhesives, <strong>and</strong> paints) due to their good durability<br />
<strong>and</strong> availability at low cost. In these applications a<br />
continuous film is formed upon drying <strong>of</strong> the latex. The<br />
mechanical properties <strong>of</strong> the polymer film depend on the<br />
molecular properties (e.g., molecular weight distribution,<br />
copolymer composition, etc.) <strong>and</strong> morphological properties<br />
(e.g., particle size distribution) <strong>of</strong> the latex.<br />
<strong>Copolymerization</strong> <strong>of</strong> vinyl acetate with other vinyl<br />
monomers can lead to the production <strong>of</strong> latex particles<br />
having a wide range <strong>of</strong> molecular <strong>and</strong> particle-morphological<br />
properties depending on the molecular structure <strong>of</strong><br />
the comonomer <strong>and</strong> the copolymer composition. One <strong>of</strong><br />
the most important industrial latexes, widely utilized in<br />
the architectural coatings market, is the vinyl acetate<br />
(VAc)/butyl acrylate (BuA) emulsion copolymer with a<br />
BuA composition <strong>of</strong> 15–25%. The emulsion copolymerization<br />
<strong>of</strong> the VAc/BuA system is characterized by large<br />
differences in reactivity ratios (r VAc = 0.05 <strong>and</strong> r BuA =<br />
5.5), water solubilities (25 g/L for VAc <strong>and</strong> 1–1.5 g/L for<br />
BuA), propagation rate constants (k p (VAc) = 4000 L/<br />
mol/s <strong>and</strong> k p (BuA) = 200 L/mol/s), <strong>and</strong> glass transition<br />
temperatures (T g (VAc) = 328C <strong>and</strong> T g (BuA) = –548C). [1]<br />
Due to these significantly different monomer properties,<br />
latexes having a wide range <strong>of</strong> molecular <strong>and</strong> particlemorphological<br />
properties can be obtained by varying the<br />
comonomer composition, thus influencing the glass transition<br />
temperature <strong>and</strong> the minimum film-forming temperature<br />
<strong>of</strong> the latex paints.<br />
The type <strong>of</strong> copolymerization process (semi-batch or<br />
batch) is also an important factor affecting the final latex<br />
[2, 3]<br />
properties. In batch copolymerization <strong>of</strong> BuA with<br />
Macromol. Chem. Phys. 2001, 202, No. 12 i WILEY-VCH Verlag GmbH, D-69451 Weinheim 2001 1022-1352/2001/1208–2614$17.50+.50/0
<strong>Semi</strong>-<strong>Batch</strong> <strong>Emulsion</strong> <strong>Copolymerization</strong> <strong>of</strong> <strong>Vinyl</strong> <strong>Acetate</strong> ... 2615<br />
VAc, BuA is depleted very rapidly due to its high reactivity<br />
ratio, while the polymerization rate <strong>of</strong> VAc prominently<br />
increases after the consumption <strong>of</strong> BuA. [1] Generally,<br />
this leads to heterogeneous particle structures with a<br />
s<strong>of</strong>t core, rich in the more hydrophobic butyl acrylate,<br />
<strong>and</strong> a harder hydrophilic shell, rich in the s<strong>of</strong>ter vinyl<br />
acetate. On the other h<strong>and</strong>, a semi-continuous emulsion<br />
copolymerization process can produce latex particles<br />
[1, 2, 4]<br />
which are nearly homogeneous. Thus, copolymer<br />
composition control in terms <strong>of</strong> optimal monomer<br />
addition policy has widely been studied. [2, 5, 6] It has been<br />
shown that homogeneous copolymers having a desired<br />
composition can be obtained by feeding the more reactive<br />
comonomer into the reactor according to an optimal<br />
[3, 7]<br />
control law. <strong>Semi</strong>-batch polymerization can prolong<br />
the nucleation period <strong>and</strong> result in a decrease <strong>of</strong> particle<br />
growth rate under monomer starved conditions, leading<br />
to the production <strong>of</strong> small sized, high solid-content<br />
latexes. These latex products exhibit increased emulsion<br />
viscosity which is frequently desirable in paint<br />
formulations. Moreover, based on the structural differences<br />
<strong>of</strong> the latex particles, the semi-continuous polymerization<br />
<strong>of</strong> VAc/BuA has been found to lead to the production<br />
<strong>of</strong> latexes having improved film forming properties.<br />
[1]<br />
Another important feature in the emulsion copolymerization<br />
<strong>of</strong> VAc/BuA is the significant difference in the<br />
water solubilities <strong>of</strong> the two monomers which strongly<br />
affects the respective monomer partition coefficients in<br />
the polymer <strong>and</strong> water phases. The high water solubility<br />
<strong>of</strong> VAc indicates that water phase polymerization will be<br />
dominant at low conversions. Thus, particle nucleation<br />
will predominately occur by the homogeneous nucleation<br />
mechanism. [2] At higher monomer conversions the particle<br />
phase volume increases <strong>and</strong> gradually becomes the<br />
main locus <strong>of</strong> polymerization. [1, 8] Kinetic studies on the<br />
semi-continuous emulsion polymerization <strong>of</strong> VAc/BuA<br />
have shown that the number <strong>of</strong> radicals per particle is<br />
large <strong>and</strong>, thus, the polymerization does not follow the<br />
classical Smith-Ewart kinetics. [9] The number <strong>of</strong> radicals<br />
per particle was determined to be between 1 <strong>and</strong> 7 for<br />
BuA compositions between 20 <strong>and</strong> 100%. [1] On the other<br />
h<strong>and</strong>, in batch copolymerizations the number <strong>of</strong> radicals<br />
per particle was initially large but decreased below 0.5 at<br />
higher monomer conversions after the consumption <strong>of</strong><br />
BuA. [1]<br />
In semi-batch emulsion polymerizations the initiator<br />
can initially be charged or/<strong>and</strong> continuously fed during<br />
polymerization. Increased initiator concentrations affect<br />
not only the polymerization kinetics but also the stability<br />
<strong>of</strong> the particles by increasing the ionic strength <strong>of</strong> the<br />
continuous aqueous phase. V<strong>and</strong>ez<strong>and</strong>e <strong>and</strong> Rudin [4]<br />
observed a nearly linear relationship between the ionic<br />
strength <strong>of</strong> the aqueous phase <strong>and</strong> the latex particle surface<br />
area. Moreover, they reported that the final monomer<br />
conversion increased when a continuous initiator addition<br />
policy was employed.<br />
In general, particle stabilization is achieved by the use<br />
<strong>of</strong> anionic surfactants, which are extensively employed in<br />
many emulsion polymerization systems. However, the<br />
limited stabilizing effectiveness <strong>of</strong> anionic surfactants at<br />
high solids (e.g., A40%) <strong>and</strong> electrolyte concentrations<br />
has led to the development <strong>of</strong> alternative stabilization<br />
techniques, including the combination <strong>of</strong> anionic <strong>and</strong><br />
nonionic surfactants. [10] VAc/BuA latex particles produced<br />
in batch reactors can have a core-shell morphology.<br />
Such core-shell latex particles are difficult to stabilize<br />
due to poor surfactant adsorption characteristics <strong>of</strong><br />
the hydrophilic VAc surface. [11] This problem appears<br />
even with the production <strong>of</strong> homogeneous particles in<br />
semi-batch reactors <strong>and</strong> has led to the development <strong>of</strong><br />
alternative stabilization methods. These include the use<br />
<strong>of</strong> functional comonomers containing weak or strong acid<br />
groups (e.g., acrylic acid), the use <strong>of</strong> steric stabilizers [12]<br />
(e.g., PVOH, HEC, PEG) <strong>and</strong> the use <strong>of</strong> polymerizable<br />
surfactants. [13, 14] Unfortunately, the use <strong>of</strong> alternative stabilization<br />
methods has <strong>of</strong>ten led to the appearance <strong>of</strong><br />
other problems (e.g., deterioration <strong>of</strong> film formation<br />
properties). As a result, there is a persisting need for the<br />
development <strong>of</strong> new surfactants for stabilization <strong>of</strong> high<br />
solids-content latexes.<br />
A limited number <strong>of</strong> publications, dealing with the use<br />
<strong>of</strong> nonionic surfactants for latex stabilization, have<br />
appeared in the open literature. [12] In general, nonionic<br />
surfactants provide stabilization that is lower than that<br />
obtained by anionic surfactant systems, <strong>of</strong>ten leading to<br />
the formation <strong>of</strong> multi-modal <strong>and</strong> broad particle size distributions.<br />
[10] V<strong>and</strong>ez<strong>and</strong>e <strong>and</strong> Rudin [4] described a seeded<br />
semi-batch emulsion polymerization process for the production<br />
<strong>of</strong> high solid VAc/BuA latexes, using the anionic<br />
surfactant sodium dodecyl sulfate, (SDS). The production<br />
<strong>of</strong> high solid-content latexes (e.g., up to 55–60%) could<br />
only be achieved by employing a nonionic surfactant in<br />
addition to SDS. However, V<strong>and</strong>ez<strong>and</strong>e <strong>and</strong> Rudin<br />
observed a reduction in the polymerization rate in the<br />
presence <strong>of</strong> a nonionic surfactant, which was attributed to<br />
the inhibition <strong>of</strong> oligomer radical entry rate by the viscous<br />
surfactant layer. In a similar study, Bataille et al. [9]<br />
employed polyoxyethylene-b-polypropylene nonionic<br />
surfactants with limited success.<br />
In the present work, a new generation <strong>of</strong> nonionic surfactants<br />
(e.g., alkyl polyglucosides) was employed for<br />
the production <strong>of</strong> high quality environmentally benign<br />
water-borne VAc/BuA latexes. All emulsion polymerization<br />
experiments were carried out in the presence <strong>of</strong> alkyl<br />
polyglucosides. The main goal <strong>of</strong> this work was to investigate<br />
the effect <strong>of</strong> the surfactant molecular structure <strong>and</strong><br />
concentration as well as the initiator addition policy on<br />
the polymerization kinetics, latex stability <strong>and</strong> particle<br />
size distribution.
2616 N. Lazaridis, A. H. Alexopoulos, C. Kiparissides<br />
Experimental Part<br />
Materials<br />
<strong>Vinyl</strong> acetate (Aldrich, +99%, 3–5 ppm MEHQ) <strong>and</strong> butyl<br />
acrylate (Aldrich, +99%, 10–55 ppm MEHQ), were used as<br />
received without any further purification. Ammonium persulfate<br />
(Merck, 98%) was used as initiator, sodium bicarbonate<br />
(Merck) as buffer <strong>and</strong> hydroquinone (Merck, +99%) as<br />
inhibitor to quench the polymerization. The nonionic surfactants<br />
(e.g., oligomeric alkyl polyglucosides) were supplied<br />
by Akzo-Nobel. They had a very low solubility in the<br />
organic phase while their solubility in the aqueous phase<br />
exhibited a small temperature dependence. The adsorption<br />
<strong>and</strong> particle stabilization properties <strong>of</strong> the surfactants<br />
depended on the number <strong>of</strong> hydrophilic glucoside segments,<br />
N S , <strong>and</strong> hydrophobic alkyl segments, N C (see Table 1). The<br />
general chemical formula <strong>of</strong> a straight-chain alkyl polyglucoside<br />
can be written as: H1[C 6 O 5 H 10 ] Ns 1O1[CH 2 ] Nc 1H.<br />
Notice that as N S increases or N C decreases, the hydrophile to<br />
lipophile balance (HLB), which is proportional to the (N S /<br />
N C ) ratio, increases (Table 1).<br />
Table 1.<br />
Surfactant<br />
Alkyl polyglucoside surfactant properties.<br />
CMC<br />
g=L<br />
N S<br />
Hydrophilic<br />
segments<br />
N C<br />
Hydrophobic<br />
segments<br />
N C /N S<br />
Ratio<br />
HLB<br />
S1 0.8 1.9 9.1 4.8 13.6<br />
S2 0.08 1.9 12/14 6.3 12.1<br />
S3 1.8 4.0 9.1 2.3 14.4<br />
S4 1.6 8 5.0 13.3<br />
S5 3.9 8 2.1<br />
Polymerization Process<br />
<strong>Emulsion</strong> copolymerization experiments were carried out in<br />
a 500 ml jacketed glass reactor (Normschliff Gerätebau<br />
Wertheim) equipped with a condenser <strong>and</strong> a nitrogen purge<br />
line. Two streams consisting <strong>of</strong> the monomer <strong>and</strong> initiator<br />
solutions were fed into the reactor using two piston pumps.<br />
The reaction mixture was thermostated to within l0.058C<br />
with the aid <strong>of</strong> a constant temperature bath (Julabo F32 HC)<br />
provided with an external temperature sensor. The reactor<br />
was equipped with a six-blade impeller. The agitation rate<br />
was set equal to 150 rpm to provide adequate mixing <strong>and</strong><br />
minimize coagulum formation.<br />
The emulsion copolymerization <strong>of</strong> VAc/BuA (80:20) was<br />
carried out at 808C following a st<strong>and</strong>ard recipe. Initially, the<br />
surfactant/buffer aqueous solution (2 wt.-% with respect to<br />
the total mass <strong>of</strong> monomers) was introduced to the reactor<br />
<strong>and</strong> thermostated at 808C while being purged with nitrogen<br />
for half an hour. Subsequently, a specified amount <strong>of</strong> the<br />
initiator dissolved in water was added to the surfactant solution.<br />
During the following four hours, the initiator <strong>and</strong> monomer<br />
solutions were pumped at constant rates into the reactor.<br />
According to our st<strong>and</strong>ard recipe, the amount <strong>of</strong> initiator<br />
initially charged as well as the total amount <strong>of</strong> initiator continuously<br />
fed into the reactor were 0.25 wt.-% on total monomers<br />
while the amount <strong>of</strong> surfactant added was 2 wt.-% on<br />
total monomers. The monomer <strong>and</strong> initiator solutions were<br />
also purged with nitrogen prior to the polymerization process<br />
for about half an hour.<br />
Instantaneous Monomer Conversion <strong>and</strong> Latex Particle Size<br />
Measurements<br />
Latex samples <strong>of</strong> about 1 ml were withdrawn from the reactor<br />
in half-hour intervals <strong>and</strong> mixed immediately with a 0.02<br />
wt.-% aqueous hydroquinone solution at a 9:1 w/w ratio <strong>and</strong><br />
then placed in an ice bath. The total solid-content was determined<br />
gravimetrically using an IR-moisture analyzer (Sartorius<br />
MA40). Thermogravimetric analysis (TGA) measurements<br />
were also employed to verify the accuracy <strong>of</strong> the <strong>of</strong><br />
the IR results. The instantaneous conversion <strong>of</strong> the two<br />
monomers at time t, defined by the ratio <strong>of</strong> the polymer mass<br />
over the total mass <strong>of</strong> the monomers fed to the reactor up to<br />
time t, was determined from the measurement <strong>of</strong> the total<br />
mass <strong>of</strong> solids. The rate <strong>of</strong> polymerization was calculated<br />
from the differentiation <strong>of</strong> the conversion versus time data.<br />
Finally, the overall monomer conversion was defined as the<br />
ratio <strong>of</strong> the polymer mass over the total mass <strong>of</strong> the monomers<br />
in the st<strong>and</strong>ard recipe.<br />
The latex samples were first diluted in deionized water to<br />
remove the monomer from the swollen particles <strong>and</strong>, then,<br />
the particle size distribution <strong>of</strong> the unswollen particles was<br />
measured by photon correlation spectroscopy (Malvern<br />
Autosizer Lo-C). The instrument covered a size range from<br />
30 to 3000 nm <strong>and</strong> was equipped with a 4 MW internal laser<br />
at 670 nm <strong>and</strong> a 64 channel digital correlator. It should be<br />
noted that the error in the experimentally measured values <strong>of</strong><br />
instantaneous monomer conversion <strong>and</strong> average particle diameter<br />
was less than l10%.<br />
Results <strong>and</strong> Discussion<br />
Effect <strong>of</strong> the Molecular Structure <strong>of</strong> the Nonionic<br />
Surfactant<br />
The molecular structure <strong>of</strong> the nonionic surfactants is<br />
characterized by the number <strong>of</strong> repeating segments <strong>of</strong> the<br />
hydrophilic <strong>and</strong> hydrophobic moieties (Table 1). These<br />
surfactant characteristics influence the physical properties<br />
such as surface coverage, surfactant volume fraction <strong>and</strong><br />
thickness <strong>of</strong> the adsorbed surfactant layer, which are crucial<br />
for the latex particle stabilization.<br />
The VAc/BuA emulsion copolymerization investigated<br />
in this work presented certain similarities to the results <strong>of</strong><br />
Piirma [15] who studied the emulsion polymerization <strong>of</strong><br />
styrene <strong>and</strong> MMA using a series <strong>of</strong> short-chain blockcopolymers<br />
(e.g., PS-b-PEO <strong>and</strong> PMMA-b-PEO) as stabilizers.<br />
For the styrene emulsion polymerization, a fourfold<br />
increase in the length <strong>of</strong> the adsorbing hydrophobic<br />
chain segment caused severe particle destabilization,<br />
resulting in a three-fold increase in the particle diameter.<br />
For the MMA emulsion polymerization, it was found that<br />
an optimum ratio <strong>of</strong> the adsorbing hydrophobic to stabilizing<br />
hydrophilic segments existed for maximum stabilization<br />
<strong>of</strong> particles. Particle destabilization occurred in the
<strong>Semi</strong>-<strong>Batch</strong> <strong>Emulsion</strong> <strong>Copolymerization</strong> <strong>of</strong> <strong>Vinyl</strong> <strong>Acetate</strong> ... 2617<br />
Figure 1. The effect <strong>of</strong> surfactant structure on (a) the instantaneous<br />
monomer conversion <strong>and</strong> (b) the average particle size<br />
(surfactant concentration 1 wt.-%; initiator addition policy<br />
0.25–0.25 wt.-%).<br />
presence <strong>of</strong> polymeric stabilizers with a high PEO content<br />
(e.g., a high ratio <strong>of</strong> hydrophilic/hydrophobic moieties)<br />
due to the decrease <strong>of</strong> surface coverage. On the<br />
other h<strong>and</strong>, particle destabilization was also observed at a<br />
low PEO content <strong>of</strong> polymeric stabilizers <strong>and</strong> was attributed<br />
to the insufficient amount <strong>of</strong> stabilizing ethylene<br />
oxide segments per adsorbed surface area. [15] Furthermore,<br />
an increase in the surfactant molecular weight<br />
resulted in a shift in the optimum value <strong>of</strong> the hydrophilic/hydrophobic<br />
chain length ratio <strong>and</strong> gave rise to a<br />
slight increase <strong>of</strong> the average particle size <strong>and</strong> polymerization<br />
rate.<br />
Figure 1a <strong>and</strong> 1b illustrate the effect <strong>of</strong> the molecular<br />
structure <strong>of</strong> the surfactant on the instantaneous monomer<br />
conversion <strong>and</strong> average particle size <strong>of</strong> latexes produced<br />
using the st<strong>and</strong>ard recipe. As can be seen in Figure 1a,<br />
the polymerization initially proceeds under non-starved<br />
conditions. After an approximate polymerization time <strong>of</strong><br />
90–120 min, depending on the surfactant type, the polymerization<br />
continues at an almost maximum rate (e.g.,<br />
the instantaneous monomer conversion is higher than<br />
80%). It should be noted that surfactants S2 <strong>and</strong> S3<br />
resulted in higher polymerization rates than that <strong>of</strong> surfactant<br />
S1 (Figure 1a). The lower polymerization rate<br />
observed for surfactant S1 was attributed to the reduced<br />
oligomer radical entry rate through the “viscous layer” <strong>of</strong><br />
the adsorbed nonionic surfactants. [4] A similar behavior<br />
has been observed for mixed surfactant systems in which<br />
the monomer conversion was found to be lower than that<br />
obtained with simple anionic surfactants even though the<br />
former provided better stabilization <strong>and</strong> an increased particle<br />
number. [4]<br />
The effect <strong>of</strong> the surfactant molecular structure on the<br />
latex particle size was found to be significant (Figure<br />
1b). The final average particle size varied by more than<br />
100 nm depending on the surfactant structure (e.g., the<br />
values <strong>of</strong> N C <strong>and</strong> N S ). It is noteworthy to notice that surfactant<br />
S1, despite having a shorter adsorbing chain segment,<br />
N C , than surfactant S2 <strong>and</strong> a shorter stabilizing<br />
chain, N S , than surfactant S3, provided the most effective<br />
particle stabilization. Assuming that surfactants S1 <strong>and</strong><br />
S2 have similar surface coverages, the observed increase<br />
in particle stabilization in the presence <strong>of</strong> surfactant S1<br />
can be attributed to the larger number <strong>of</strong> stabilizing<br />
chains per unit adsorbed area. On the other h<strong>and</strong>, because<br />
<strong>of</strong> the substantially different values <strong>of</strong> the hydrophobic/<br />
hydrophilic segment ratios for surfactants S1 <strong>and</strong> S3<br />
(e.g., 4.5 <strong>and</strong> 2.3, respectively) the surface coverage <strong>of</strong><br />
surfactant S3 will be lower than that <strong>of</strong> surfactant S1,<br />
resulting in a reduced particle stabilization ability.<br />
The above arguments were confirmed by comparison<br />
<strong>of</strong> the experimental data to the predictions <strong>of</strong> a comprehensive<br />
mathematical emulsion copolymerization model<br />
that included the effect <strong>of</strong> steric stabilization. [16] The<br />
values <strong>of</strong> the saturated surface coverages were obtained<br />
on the basis <strong>of</strong> best fit <strong>of</strong> theoretical stabilization model<br />
predictions to experimental data. It was found that the<br />
surfactant volume fraction <strong>of</strong> the adsorbed layer, f, varied<br />
with the hydrophobic/hydrophilic segment ratio, N C /N S ,<br />
according to the following expression:<br />
u ¼ N C<br />
N S<br />
<br />
0:107 0:0111 N <br />
C<br />
0:135<br />
N S<br />
ð1Þ<br />
Notice that for a value <strong>of</strong> (N C /N S ) ratio <strong>of</strong> 4.8, Equation<br />
(1) exhibits a maximum value <strong>of</strong> 0.123 with respect to the<br />
volume fraction u. Furthermore, it was found that the diffusional<br />
radical entry rate to the particles increased with a<br />
decrease <strong>of</strong> the volume fraction <strong>of</strong> the adsorbed layer, u. [16]<br />
Based on the above observations, surfactant S1 will provide<br />
the best stabilization, for it has the largest value <strong>of</strong> u<br />
<strong>and</strong> exhibits the lowest polymerization rate due to the largest<br />
“viscous layer” resistance to the radical entry rate.<br />
Effect <strong>of</strong> Surfactant Concentration<br />
Figure 2a <strong>and</strong> 2b illustrate the effect <strong>of</strong> concentration <strong>of</strong><br />
surfactant S1 on the instantaneous conversion <strong>and</strong> the
2618 N. Lazaridis, A. H. Alexopoulos, C. Kiparissides<br />
Figure 4. The effect <strong>of</strong> surfactant concentration on the average<br />
particle size: (surfactant S3; initiator addition policy: 0.25–0.25<br />
wt.-%).<br />
Figure 2. The effect <strong>of</strong> surfactant concentration on (a) the<br />
instantaneous monomer conversion <strong>and</strong> (b) the average particle<br />
size (surfactant S1; initiator addition policy: 0.25–0.25 wt.-%).<br />
Figure 3. The effect <strong>of</strong> surfactant concentration on the average<br />
particle size (surfactant S2; initiator addition policy: 0.25–0.25<br />
wt.-%).<br />
average particle size. It can be seen that as the concentration<br />
<strong>of</strong> surfactant S1 increases from 0.03 to 1.8 wt.-% the<br />
polymerization rate increases (Figure 2a) while the particle<br />
stabilization is improved (Figure 2b). However, at<br />
very high surfactant concentrations (e.g., 3.7 wt.-% on<br />
total monomers) particle destabilization <strong>and</strong> lower polymerization<br />
rates were experienced. Particle destabilization<br />
at high surfactant concentrations was also observed<br />
for surfactant S2 but to a smaller extent (Figure 3). It is<br />
important to point out that no particle destabilization was<br />
observed in the presence <strong>of</strong> surfactant S3 even at high<br />
concentrations (Figure 4). The particle destabilization<br />
observed at high concentrations <strong>of</strong> surfactant S1 can be<br />
attributed to a depletion mechanism caused by displaced<br />
surfactant molecules or micelles that may trigger the<br />
nucleation <strong>of</strong> a second generation <strong>of</strong> particles, leading to<br />
small-large particle agglomeration.<br />
The effect <strong>of</strong> surfactant concentration on the evolution<br />
<strong>of</strong> the particle size distribution (PSD) is illustrated in Figure<br />
5. It can be seen that the PSD generally evolves from<br />
a bimodal distribution to a unimodal one. The bimodal<br />
distribution at low conversions is a manifestation <strong>of</strong> the<br />
combined action <strong>of</strong> the micellar <strong>and</strong> homogeneous<br />
nucleation mechanisms. When the surfactant concentration<br />
is above the CMC (see Figure 5b <strong>and</strong> 5c) the PSD<br />
initially exhibits a bimodal form. The first peak <strong>of</strong> the<br />
distribution at 50–60 nm can be attributed to micellar<br />
nucleation <strong>of</strong> particles while the second peak at 65–<br />
75 nm can be related with the homogeneous particle<br />
nucleation mechanism. Because <strong>of</strong> the increased stability<br />
<strong>of</strong> latex particles formed by micellar nucleation, the first<br />
peak <strong>of</strong> the distribution is narrower <strong>and</strong> includes smallersize<br />
particles than the homogeneous nucleation peak. The<br />
two peaks gradually merge together with time due to particle<br />
coalescence. It should be pointed out that the most<br />
stable <strong>and</strong> narrowest PSD was obtained at an intermediate<br />
surfactant concentration (Figure 5b). On the other h<strong>and</strong>,<br />
at large surfactant concentrations (Figure 5c) particle<br />
destabilization occurred while the bimodal character <strong>of</strong><br />
the distribution was retained for longer polymerization<br />
times, leading eventually to broader final PSD’s (Figure<br />
5c).<br />
Effect <strong>of</strong> Initiator Addition Policy<br />
The effect <strong>of</strong> initiator addition policy on the instantaneous<br />
monomer conversion <strong>and</strong> the average particle size<br />
was also investigated. In the experiments carried out, the
<strong>Semi</strong>-<strong>Batch</strong> <strong>Emulsion</strong> <strong>Copolymerization</strong> <strong>of</strong> <strong>Vinyl</strong> <strong>Acetate</strong> ... 2619<br />
Figure 6. The effect <strong>of</strong> initiator addition policy on (a) the<br />
instantaneous monomer conversion <strong>and</strong> (b) the average particle<br />
size (surfactant S1 concentration 2 wt.-%).<br />
Figure 5. The effect <strong>of</strong> surfactant concentration on the particle<br />
size distribution. Surfactant S1 concentration: (a) 0.06, (b) 0.9,<br />
<strong>and</strong> (c) 1.8 wt.-% to monomers (initiator addition policy: 0.25–<br />
0.25 wt.-%).<br />
initial amount <strong>of</strong> initiator or the amount <strong>of</strong> initiator continuously<br />
fed to the reactor was varied. Figure 6a <strong>and</strong> 6b<br />
depict the time evolution <strong>of</strong> the instantaneous monomer<br />
conversion <strong>and</strong> average particle size for various initiator<br />
addition policies. Notice that the rate <strong>of</strong> polymerization<br />
<strong>and</strong> the final monomer conversion increase as the total<br />
amount <strong>of</strong> initiator added to the reactor (e.g., initially<br />
charged <strong>and</strong> continuously fed) increases up to the total<br />
recipe value <strong>of</strong> 0.5 wt.-%. An increase in the initiator<br />
amount initially charged or/<strong>and</strong> in the initiator amount<br />
continuously fed to the reactor above the respective<br />
recipe values (e.g., <strong>of</strong> 0.25 wt.-% initially charged <strong>and</strong><br />
0.25 wt.-% continuously fed) led to severe destabilization<br />
<strong>and</strong> reduction <strong>of</strong> the final monomer conversion.<br />
It was observed that when the initiator amount initially<br />
charged was increased above its recipe value <strong>of</strong> 0.25<br />
wt.-%, destabilization occurred early in the polymerization<br />
(Figure 6b). On the other h<strong>and</strong>, when the initiator<br />
amount continuously fed was increased above its recipe<br />
value, destabilization occurred later in the reaction. In all<br />
other cases, the effect <strong>of</strong> charged <strong>and</strong> fed initiator<br />
amounts could not be distinguished as evidenced by the<br />
experimental results obtained with 0.12 wt.-% initially<br />
charged <strong>and</strong> 0.13 wt.-% continuously fed initiator<br />
amounts (see Figure 6a <strong>and</strong> 6b). The extent <strong>of</strong> particle<br />
destabilization caused by an increase in the initiator concentration<br />
was less significant in the presence <strong>of</strong> surfactant<br />
S2 (Figure 7) <strong>and</strong> insignificant for surfactant S3 (Figure<br />
8).<br />
The time evolution <strong>of</strong> the PSD at constant surfactant<br />
concentration <strong>and</strong> different initiator addition policies is<br />
depicted in Figure 9. The bimodal to unimodal evolution
2620 N. Lazaridis, A. H. Alexopoulos, C. Kiparissides<br />
Figure 7. The effect <strong>of</strong> the initial initiator concentration on the<br />
average particle size (surfactant S2: 1 wt.-%).<br />
Figure 8. The effect <strong>of</strong> the initial initiator concentration on the<br />
average particle size (surfactant S3: 1 wt.-%).<br />
<strong>of</strong> PSD which was observed with the nominal initiator<br />
addition policy (Figure 9a), was also observed for the<br />
case <strong>of</strong> an increased initiator addition policy (e.g., 0.45<br />
wt.-% initially charged <strong>and</strong> 0.25 wt.-% continuously fed)<br />
(Figure 9c) but led to a narrower PSD located at larger<br />
particle sizes. However, when the amount <strong>of</strong> initiator<br />
continuously fed increased from 0.25 wt.-% to 0.53<br />
wt.-%, the PSD remained bimodal <strong>and</strong> evolved to a<br />
multi-modal PSD located at larger particle sizes (Figure<br />
9b).<br />
Other investigators [4] have reported a similar reduction<br />
in the average particle size as a result <strong>of</strong> a decrease in the<br />
initiator concentration in the presence <strong>of</strong> anionic or/<strong>and</strong><br />
cationic surfactants. This behavior was attributed to the<br />
decrease <strong>of</strong> the ionic strength <strong>of</strong> the reaction medium<br />
with the concomitant improvement <strong>of</strong> the stability <strong>of</strong> the<br />
primary particles. It should be noted that even with nonionic<br />
surfactants the ionic strength could influence the<br />
steric stabilization <strong>of</strong> the particles by changing either the<br />
thickness <strong>of</strong> the adsorbed surfactant layer or the surfactant<br />
surface coverage.<br />
In order to elucidate whether or not the ionic strength<br />
<strong>of</strong> the reaction medium was the main mechanism for the<br />
Figure 9. The effect <strong>of</strong> initiator addition policy on the particle<br />
size distribution (surfactant S1, 1 wt.-%; initiator addition policy<br />
(a) 0.25–0.25 wt.-%, (b) 0.25–0.53 wt.-%, (c) 0.45–0.25<br />
wt.-%).<br />
observed destabilization <strong>of</strong> latex particles at high initiator<br />
concentrations, three experiments were carried out by<br />
varying the initiator <strong>and</strong> electrolyte concentrations but<br />
keeping the total ionic strength <strong>of</strong> the reaction medium<br />
constant. The ionic strength <strong>of</strong> the medium, I, was calculated<br />
based on the concentrations <strong>of</strong> the ionic species <strong>of</strong><br />
initiator (1:2) (C I : Na 2 S 2 O 8 ), <strong>and</strong> those <strong>of</strong> electrolyte<br />
(1:1) (C E : NaHCO 3 ). Accordingly, the total ionic strength<br />
<strong>of</strong> the reaction medium will be equal to: I = 3 N C I + C E . It
<strong>Semi</strong>-<strong>Batch</strong> <strong>Emulsion</strong> <strong>Copolymerization</strong> <strong>of</strong> <strong>Vinyl</strong> <strong>Acetate</strong> ... 2621<br />
Figure 10. The effect <strong>of</strong> initial initiator <strong>and</strong> electrolyte concentrations<br />
at constant ionic strength on the average particle size<br />
(surfactant S2: 1 wt.-%; 0.25 wt.-% initiator continuously fed).<br />
was found that even at constant ionic strength, differences<br />
in the average particle size were observed (Figure 10)<br />
implying a possible interaction mechanism between<br />
initiator <strong>and</strong> surfactant, which influences the latex particle<br />
stabilization or/<strong>and</strong> the radical entry rate. The most<br />
stable case was observed when using a low initiator concentration<br />
<strong>and</strong> a high electrolyte concentration (e.g., C I =<br />
0.013 mol/L <strong>and</strong> C E = 0.023 mol/L electrolyte). Notice<br />
that increased initiator concentrations eventually lead to<br />
larger particle sizes.<br />
Effect <strong>of</strong> Monomer Addition Rate<br />
The existence <strong>of</strong> “pseudo-steady states” in semi-batch<br />
emulsion copolymerization has been investigated <strong>and</strong><br />
reported in the literature for various polymerization systems.<br />
[5, 6, 17–19] According to these reports, there is a critical<br />
monomer addition rate above which the monomer concentration<br />
in the latex particles becomes saturated. Thus,<br />
above this critical value, the polymerization rate remains<br />
constant irrespectively <strong>of</strong> the monomer addition rate provided<br />
that the monomer diffusion rate through the aqueous<br />
phase is larger than the polymerization rate. On the<br />
other h<strong>and</strong>, for monomer addition rates below the critical<br />
addition rate, the polymerization rate will be equal to the<br />
monomer addition rate. The existence <strong>of</strong> the critical<br />
monomer addition rate has been experimentally tested<br />
<strong>and</strong> confirmed with monomers having different water<br />
solubilities such as styrene <strong>and</strong> vinyl acetate.<br />
In the present study, the effect <strong>of</strong> monomer addition<br />
rate was examined using surfactant S3. It was observed<br />
that the polymerization rate was initially increased (e.g.,<br />
in the first 30–60 min) <strong>and</strong> then reached a constant value<br />
which was equal to the respective monomer addition rate<br />
(Figure 11a). The small overshoot observed in the polymerization<br />
rate at about 90 min was attributed to the<br />
polymerization <strong>of</strong> the excess monomer accumulated in<br />
the aqueous phase during the non-starved period (Figure<br />
Figure 11. The effect <strong>of</strong> monomer addition rate on (a) the<br />
polymerization rate <strong>and</strong> (b) the average particle size. Dashed<br />
lines indicate the monomer addition rates (surfactant S3: 2<br />
wt.-%; initiator addition policy 0.25–0.25 wt.-%).<br />
11a). Furthermore, it was observed that an increase in the<br />
monomer addition rate resulted in an increase in the average<br />
particle size (Figure 11b). Notice that larger monomer<br />
addition rates result in higher monomer concentrations<br />
in the latex particles <strong>and</strong>, thus, in larger particle<br />
growth rates. The above results for the polymerization<br />
rate (Figure 11a) <strong>and</strong> the average particle size (Figure<br />
11b) are in agreement with the results <strong>of</strong> Dimitratos et<br />
al. [5, 6] obtained for the semi-batch emulsion copolymerization<br />
<strong>of</strong> VAc/BuA (80:20) at 608C.<br />
Conclusions<br />
For all the investigated alkyl polyglucoside surfactants, it<br />
was found that the latex particle stability <strong>and</strong> the rate <strong>of</strong><br />
polymerization increased with an increase in the surfactant<br />
concentration up to the st<strong>and</strong>ard recipe value. However,<br />
excessive surfactant concentrations resulted in particle<br />
destabilization. As a result, the final average particle<br />
size exhibited a “U-shape” behavior with respect to the<br />
surfactant concentration. A possible cause for the<br />
observed particle destabilization at high surfactant con-
2622 N. Lazaridis, A. H. Alexopoulos, C. Kiparissides<br />
centrations may be attributed to bridging or/<strong>and</strong> micellar<br />
depletion flocculation. Particle destabilization was also<br />
observed when either the fed continuously or/<strong>and</strong> initially<br />
charged initiator amounts were increased above the<br />
respective recipe values. The latex particle size distributions<br />
usually evolved from bimodal to unimodal shapes.<br />
Furthermore, it was shown that latex particle stabilization<br />
significantly depended on the surfactant structure.<br />
The key factor for imparting stability was the hydrophilic/hydrophobic<br />
chain length ratio <strong>of</strong> the APG surfactants<br />
represented by the segment number ratio N S /N C . For small<br />
values <strong>of</strong> the N S /N C ratio, the density <strong>of</strong> the stabilizing<br />
chains in the adsorbed surfactant layer was insufficient to<br />
provide stabilization while large values <strong>of</strong> N S /N C led to a<br />
decreased surfactant surface coverage. Thus, optimum<br />
particle stabilization could be achieved at a specific<br />
hydrophilic/hydrophobic chain length ratio. The results <strong>of</strong><br />
the present investigation provide useful guidelines for<br />
choosing the best surfactant from an homologous series<br />
<strong>of</strong> APG’s for optimum latex particle stabilization.<br />
Acknowledgement: The authors gratefully acknowledge the<br />
DGXII <strong>of</strong> EU for supporting this work under the BRITE/EURAM<br />
Project BE 95-1214.<br />
Received: August 2, 2000<br />
Revised: December 8, 2000<br />
[1] X. Kong, C. Pichot, J. Guillot, Eur. Polym. J. 1988, 24,<br />
485.<br />
[2] M. El Aasser, T. Makgawinata, J. V<strong>and</strong>erh<strong>of</strong>f, C. Pichot, J.<br />
Polym. Sci., Polym. Chem. 1983, 21, 2363.<br />
[3] S. Misra, C. Pichot, M. El Aasser, J. V<strong>and</strong>erh<strong>of</strong>f, J. Polym.<br />
Sci., Polym. Chem. 1983, 21, 2383.<br />
[4] G. V<strong>and</strong>erz<strong>and</strong>e, A. Rudin, in: “Polymer Latexes: Preparation,<br />
Characterization <strong>and</strong> Applications”, ACS Symposium<br />
Series, 1992, Chapters 8 <strong>and</strong> 9.<br />
[5] J. Dimitratos, C. Georgakis, M. El Aasser, A. Klein, Comput.<br />
Chem. Eng. 1989, 13, 21.<br />
[6] J. Dimitratos, M. El Aasser, C. Georgakis, A. Klein, J.<br />
Appl. Polym. Sci. 1990, 40, 1005.<br />
[7] K. Chujo, Y. Harada, S. Tokuhara, K. Tanaka, J. Polym.<br />
Sci. C 1969, 27, 321.<br />
[8] I. Capek, J. Barton, Makromol. Chem. 1985, 186, 1297.<br />
[9] P. Bataille, B. T. Van, Q. B. Pham, J. Appl. Polym. Sci.<br />
1978, 22, 3145.<br />
[10] E. Unzueta, J. Forcada, Polymer 1995, 36, 1045.<br />
[11] B. Vijayendran, in: “Polymer Colloids II”, R. Fitch, Ed.,<br />
Plenum Press, 1980.<br />
[12] M. J. Westby, Colloid Polym. Sci. 1988, 266, 46.<br />
[13] M. B. Urquiola, V. L. Dimonie, E. D. Sudol, M. S. El Aasser,<br />
J. Polym. Sci., Polym. Chem. 1992, 30, 2612.<br />
[14] M. B. Urquiola, V. L. Dimonie, E. D. Sudol, M. S. El Aasser,<br />
J. Polym. Sci., Polymer Chem. 1992, 30, 2631.<br />
[15] I. Piirma, “Polymeric Surfactants”, Marcel Dekker, Inc.,<br />
1992, p. 138–146.<br />
[16] N. Lazarides, A. H. Alexopoulos, E. G. Chatzi, C. Kiparissides,<br />
Chem. Eng. Sci. 1999, 54, 3251.<br />
[17] H. Gerrens, J. Polym. Sci. Part C 1969, 27, 77.<br />
[18] R. A. Wessling, J. Appl. Polym. Sci. 1968, 12, 309.<br />
[19] J. Snuparek, F. Krska, J. Appl. Polym. Sci. 1976, 20, 1753.














