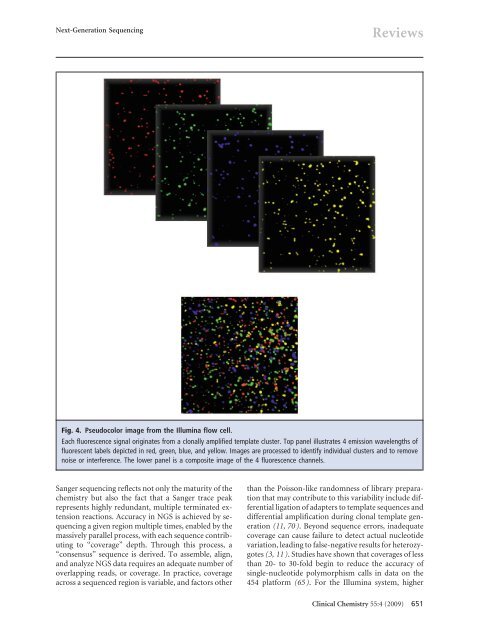
Reviewsined the genomic-binding sites of the human NRSF(neuron restrictive silencer fac<strong>to</strong>r) and STAT1 (signaltransducer and activa<strong>to</strong>r of transcription 1) proteinsindicate the resolution of ChIP-Seq <strong>to</strong> be greater thanfor ChIP-on-chip, as evidenced by confirmation ofpreviously identified binding sites and identification ofnovel binding sites (56, 57). Analogous <strong>to</strong> RNA-Seq,ChIP-Seq has the important advantage of not requiringprior knowledge of genomic locations of protein binding.In addition <strong>to</strong> the study of transcription fac<strong>to</strong>rs,NGS is being used <strong>to</strong> map genomic methylation. Oneapproach involves traditional bisulfite conversion ofDNA followed by NGS, which has been applied <strong>to</strong> thestudy of entire genomes or genomic subregions(58, 59). Ongoing studies are attempting <strong>to</strong> develop avariant of ChIP-Seq in which genomic methylation isassayed by coupling immunoprecipitation with amonoclonal antibody directed against methylated cy<strong>to</strong>sineand subsequent NGS (60).NGS Data AnalysisNGS experiments generate unprecedented volumes ofdata, which present challenges and opportunities fordata management, s<strong>to</strong>rage, and, most importantly,analysis (61). NGS data begin as large sets of tiled fluorescenceor luminescence images of the flow-cell surfacerecorded after each iterative sequencing step (Fig.4). This volume of data requires a resource-intensivedata-pipeline system for data s<strong>to</strong>rage, management,and processing. Data volumes generated during singleruns of the 454 GS FLX, Illumina, and SOLiD instrumentsare approximately 15 GB, 1 TB, and 15 TB, respectively.The main processing feature of the datapipeline is the computationally intensive conversion ofimage data in<strong>to</strong> sequence reads, known as base calling.First, individual beads or clusters are identified and localizedin an image series. Image parameters such asintensity, background, and noise are then used in aplatform-dependant algorithm <strong>to</strong> generate read sequencesand error probability–related quality scoresfor each base. Although many researchers use the basecalls generated by the platform-specific data-pipelinesoftware, alternative base-calling programs that usemore advanced software and statistical techniqueshave been developed. Features of these alternative programsinclude the incorporation of ambiguous basesin<strong>to</strong> reads, improved removal of poor-quality basesfrom read ends (62), and the use of data sets for softwaretraining (15). Incorporation of these features hasbeen shown <strong>to</strong> reduce read error and improve alignment,especially as platforms are pushed <strong>to</strong> generatelonger reads. These advantages, however, must beweighed against the substantial computer resources requiredby the large volumes of image data.The quality values calculated during NGS basecalling provide important information for alignment,assembly, and variant analysis. Although the calculationof quality varies between platforms, the calculationsare all related <strong>to</strong> the his<strong>to</strong>rically relevant phredscore, introduced in 1998 for Sanger sequence data(63, 64). The phred score quality value, q, uses a mathematicalscale <strong>to</strong> convert the estimated probability ofan incorrect call, e, <strong>to</strong> a log scale:q 10 log 10 e.Miscall probabilities of 0.1 (10%), 0.01 (1%), and 0.001(0.1%) yield phred scores of 10, 20, and 30, respectively.The NGS error rates estimated by quality valuesdepend on several fac<strong>to</strong>rs, including signal-<strong>to</strong>-noiselevels, cross talk from nearby beads or clusters, anddephasing. Substantial effort has been made <strong>to</strong> understandand improve the accuracy of quality scores andthe underlying error sources (10, 14), including inaccuraciesin homopolymer run lengths on the 454 platformand base-substitution error biases with the Illuminaformat. Study of these error traits has led <strong>to</strong>examples of software that require no additional basecalling but that improve quality-score accuracies andthus improve sequencing accuracy (65, 66). Qualityvalues are an important <strong>to</strong>ol for rejecting low-qualityreads, trimming low-quality bases, improving alignmentaccuracy, and determining consensus-sequenceand variant calls (67).Alignment and assembly are substantially moredifficult for NGS data than for Sanger data because ofthe shorter reads lengths in the former. One limitationof short-read alignment and assembly is the inability <strong>to</strong>uniquely align large portions of a read set when the readlength becomes <strong>to</strong>o short. Similarly, the number ofuniquely aligned reads is reduced when aligning <strong>to</strong>larger, more complex genomes or reference sequencesbecause of their having a higher probability of repetitivesequences. A case in point is a modeling study thatindicated that 97% of the E. coli genome can beuniquely aligned with 18-bp reads but that only 90% ofthe human genome can be uniquely aligned with 30-bpreads (68, 69). Unique alignment or assembly is reducednot only by the presence of repeat sequences butalso by shared homologies within closely related genefamilies and pseudogenes. Nonunique read alignmentis handled in software by read distribution betweenmultiple alignment positions or leaving alignmentgaps. De novo assembly will reject these reads, leading<strong>to</strong> shorter and more numerous assembled contigs.These fac<strong>to</strong>rs are relevant when choosing an appropriatesequencing platform with its associated read length,particularly for de novo assembly (9).Error rates for individual NGS reads are higherthan for Sanger sequencing. The higher accuracy of650 Clinical Chemistry 55:4 (2009)
<strong>Next</strong>-<strong>Generation</strong> <strong>Sequencing</strong>ReviewsFig. 4. Pseudocolor image from the Illumina flow cell.Each fluorescence signal originates from a clonally amplified template cluster. Top panel illustrates 4 emission wavelengths offluorescent labels depicted in red, green, blue, and yellow. Images are processed <strong>to</strong> identify individual clusters and <strong>to</strong> removenoise or interference. The lower panel is a composite image of the 4 fluorescence channels.Sanger sequencing reflects not only the maturity of thechemistry but also the fact that a Sanger trace peakrepresents highly redundant, multiple terminated extensionreactions. Accuracy in NGS is achieved by sequencinga given region multiple times, enabled by themassively parallel process, with each sequence contributing<strong>to</strong> “coverage” depth. Through this process, a“consensus” sequence is derived. To assemble, align,and analyze NGS data requires an adequate number ofoverlapping reads, or coverage. In practice, coverageacross a sequenced region is variable, and fac<strong>to</strong>rs otherthan the Poisson-like randomness of library preparationthat may contribute <strong>to</strong> this variability include differentialligation of adapters <strong>to</strong> template sequences anddifferential amplification during clonal template generation(11, 70). Beyond sequence errors, inadequatecoverage can cause failure <strong>to</strong> detect actual nucleotidevariation, leading <strong>to</strong> false-negative results for heterozygotes(3, 11). Studies have shown that coverages of lessthan 20- <strong>to</strong> 30-fold begin <strong>to</strong> reduce the accuracy ofsingle-nucleotide polymorphism calls in data on the454 platform (65). For the Illumina system, higherClinical Chemistry 55:4 (2009) 651