Neuropathology ArticleCl<strong>in</strong>ical <strong>and</strong> Neuropathological Investigations <strong>in</strong>Creutzfeldt-Jakob DiseaseTransmissible spongiform encephalopathies (TSEs),also known as prion diseases, are a group of rare<strong>and</strong> <strong>in</strong>variably fatal degenerative diseases of thecentral nervous system affect<strong>in</strong>g humans as well as a numberof animal species. 1 Enormous public <strong>and</strong> scientificattention has focused on prion diseases, not only becauseof their unique biological properties, but also because oftheir impact on animal <strong>and</strong> public health, particularlywith the emergence of bov<strong>in</strong>e spongiform encephalopathy(BSE) 2 <strong>and</strong> variant Creutzfeldt-Jakob disease (variantCJD) <strong>in</strong> the United K<strong>in</strong>gdom. 3 Unlike other forms of CJD,<strong>in</strong>fectivity is readily detectable with<strong>in</strong> lymphoid tissues <strong>in</strong>variant CJD, 4 rais<strong>in</strong>g concerns over the potential spread ofvariant CJD by iatrogenic means, particularly throughsurgical procedures <strong>and</strong> surgical <strong>in</strong>struments, as the <strong>in</strong>fectiousagent shows an alarm<strong>in</strong>g resistance to conventionaldecontam<strong>in</strong>ation methods. More recently it has beenshown that variant CJD also appears to be transmissibleby blood transfusion, heighten<strong>in</strong>g concerns over secondaryhuman-to-human spread of the disease via contam<strong>in</strong>atedblood products. 5,6In humans, prion diseases occur <strong>in</strong> three ma<strong>in</strong> groups;they may occur sporadically, by autosomal dom<strong>in</strong>ant<strong>in</strong>heritance through mutations or <strong>in</strong>sertions <strong>in</strong> the prionprote<strong>in</strong> gene (PRNP), or by secondary transmissionthrough either dietary or medical exposure to the <strong>in</strong>fectiousagent. 7 Traditionally, human prion diseases are classifiedaccord<strong>in</strong>g to their major cl<strong>in</strong>ical features <strong>in</strong>toCreutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Sche<strong>in</strong>ker disease (GSS), fatal familial <strong>in</strong>somnia (FFI) <strong>and</strong>kuru (Table 1). All forms of prion disease share four neuropathologicalfeatures (spongiform vacuolation, neuronalloss, astrocytic <strong>and</strong> microglial proliferation <strong>and</strong> <strong>in</strong> certa<strong>in</strong>cases the presence of amyloid plaques), which althoughcharacteristic of these disorders are not entirely specific. 8All prion diseases are associated with the conversion ofthe normal cellular host encoded prion prote<strong>in</strong>, PrP C , to anabnormal disease-associated isoform, PrP SC .PrP SC is notonly a diagnostic marker of disease, but has been proposedas the sole or pr<strong>in</strong>cipal component of the transmissibleagent <strong>in</strong> prion disease. Accord<strong>in</strong>g to the ‘prion hypothesis’,PrP SC is derived from the normal cellular prote<strong>in</strong> (PrP C ) bya post-translational mechanism, which appears to <strong>in</strong>volvea conformational change. 9 This <strong>in</strong>volves refold<strong>in</strong>g of theprote<strong>in</strong> to a structure conta<strong>in</strong><strong>in</strong>g a high beta sheet content,which readily forms aggregates <strong>and</strong> is more resistant todenaturation by proteases than PrP C .Genetic <strong>and</strong> molecular aspects of sporadic CJDThe most common form of human prion disease is sporadicCJD, which accounts for around 85% of all humanprion diseases, with a world wide <strong>in</strong>cidence of around 1-Table 1: Classification of human prion diseasesAetiologySporadicFamilialAcquiredDiseaseSporadic Creutzfeldt-Jakob DiseaseSporadic Fatal InsomniaFamilial CJDGerstmann-Sträussler-Sche<strong>in</strong>kerFatal Familial InsomniaKuru (human source)Iatrogenic CJD (human source)Variant CJD (bov<strong>in</strong>e source)1.5 cases per million of the population per annum. Like allhuman prion diseases, much phenotypic heterogeneityexists with<strong>in</strong> sporadic CJD <strong>in</strong> terms of cl<strong>in</strong>ical <strong>and</strong> pathologicalfeatures. 10 This heterogeneity has been l<strong>in</strong>ked withthe polymorphism found at codon 129 on PRNP whichencodes either methion<strong>in</strong>e (M) or val<strong>in</strong>e (V). 11 This polymorphismhas also been identified as an important riskfactor <strong>in</strong> sporadic CJD; most cases occur <strong>in</strong> <strong>in</strong>dividualswho are homozygous for methion<strong>in</strong>e at codon 129, whopresent with the most ‘typical’ cl<strong>in</strong>ical <strong>and</strong> pathologicalfeatures. Cases of sporadic CJD <strong>in</strong> heterozygotes <strong>and</strong>val<strong>in</strong>e homozygotes are rarer <strong>and</strong> display more ‘atypical’phenotypes (Table 2). 12The physicochemical properties of PrP SC also play animportant role <strong>in</strong> <strong>in</strong>fluenc<strong>in</strong>g the disease phenotype <strong>in</strong>sporadic CJD. Western blot analysis of the protease resistantcore of PrP SC , referred to as PrPres, has identified twok<strong>in</strong>ds of heterogeneity with<strong>in</strong> the bra<strong>in</strong>s of patients withCJD. Firstly, differences occur <strong>in</strong> the mobility of the proteaseresistant core, presumably relat<strong>in</strong>g to differentPrPres fragment sizes after prote<strong>in</strong>ase K-mediated N-term<strong>in</strong>altruncation, <strong>and</strong> secondly, variation occurs <strong>in</strong> therelative abundance of the three PrP glycoforms (diglycosylated,monoglycosylated <strong>and</strong> nonglycosylated).Follow<strong>in</strong>g the classification of Parchi et al. 13 two dist<strong>in</strong>ctPrP SC types or PrPres isotypes, have been identified afterprote<strong>in</strong>ase K digestion: one with a mobility on westernblot of around 21kDa named PrPres type 1, <strong>and</strong> the second,which is slightly smaller with a molecular weight ofaround 19kDa named PrPres type 2 (Figure 1). 13 This classificationsystem has been further subdivided to <strong>in</strong>corporatePrPres isotype comb<strong>in</strong>ed with codon 129 PRNPgenotype (MM, MV, VV) result<strong>in</strong>g <strong>in</strong> six different sporadicCJD subtypes. 14 Exam<strong>in</strong>ation of the cl<strong>in</strong>ical <strong>and</strong>pathological data from each of these subtypes shows thatalthough not all have a dist<strong>in</strong>ct phenotype, there doesappear to be a good correlation between cl<strong>in</strong>ical <strong>and</strong> neuropathologicalfeatures <strong>and</strong> disease subtype (Table 2).More recently, the observation of CJD patients with morethan one PrPres isotype with<strong>in</strong> the bra<strong>in</strong> has comb<strong>in</strong>ed to<strong>in</strong>crease the heterogeneity <strong>and</strong> complexity observed <strong>in</strong>sporadic CJD.The presence of dist<strong>in</strong>ct stra<strong>in</strong>s of the <strong>in</strong>fectious agent<strong>in</strong> prion diseases has been established for some time, particularly<strong>in</strong> relation to scrapie <strong>in</strong> sheep. However, the presenceof <strong>in</strong>dividual stra<strong>in</strong>s rema<strong>in</strong>s difficult to expla<strong>in</strong>with<strong>in</strong> the bounds of the prion hypothesis, which proposesthat all the <strong>in</strong>formation required for <strong>in</strong>dividual stra<strong>in</strong>phenotypes is conta<strong>in</strong>ed with<strong>in</strong> the prion prote<strong>in</strong> itself. 9In sporadic CJD, the different conformations of PrPres asdeterm<strong>in</strong>ed by western blot analysis have been proposedto represent different biological profiles of the transmissibleagent, which may <strong>in</strong> turn relate to different biologicalstra<strong>in</strong>s. Confirmation that these different molecular conformationsor isoforms of the prion prote<strong>in</strong> do <strong>in</strong>deedcorrespond to dist<strong>in</strong>ct stra<strong>in</strong>s will require analysis of thebiological properties (such as <strong>in</strong>cubation period <strong>and</strong> pat-Professor James Ironside is aNeuropathologist <strong>in</strong> Ed<strong>in</strong>burghwho has been <strong>in</strong>volved <strong>in</strong> theNational CJD Surveillance Units<strong>in</strong>ce 1990. In 1996, he <strong>and</strong> his colleaguesidentified variant CJD, thenew human prion disease which isl<strong>in</strong>ked to BSE. His current research<strong>in</strong>terests <strong>in</strong>clude the tissue distributionof <strong>in</strong>fectivity <strong>in</strong> all forms ofCJD, <strong>and</strong> the neuropathology ofpaediatric gliomas.Diane Ritchie started her career <strong>in</strong>prion diseases at the Institute forAnimal Health, NeuropathogenesisUnit, under the guidance ofProfessor Moira Bruce look<strong>in</strong>g atexperimental models of scrapie.Currently she is study<strong>in</strong>g at theNational CJD Surveillance Unitlook<strong>in</strong>g at human prion diseaseswith Professor James Ironside,where she has ref<strong>in</strong>ed the PET blottechnique for use on human tissue.Correspondence to:Ms Diane L Ritchie,National CJD Surveillance Unit,Western General Hospital,Ed<strong>in</strong>burgh EH4 2XU, UK.Tel: 0131 537 1980Fax: 0131 537 3056Email: diane.ritchie@ed.ac.ukFigure 1: Western blot analysis of PrPres from the frontalcortex of two different sporadic CJD (s) patients, show<strong>in</strong>g thetype 1 <strong>and</strong> type 2 mobility variants. The dist<strong>in</strong>ctive type 2Bpattern found <strong>in</strong> variant CJD (v) patients, with apredom<strong>in</strong>ance <strong>in</strong> the diglycosylated PrPres is also shown.20 I <strong>ACNR</strong> • VOLUME 5 NUMBER 6 • JANUARY/FEBRUARY 2006
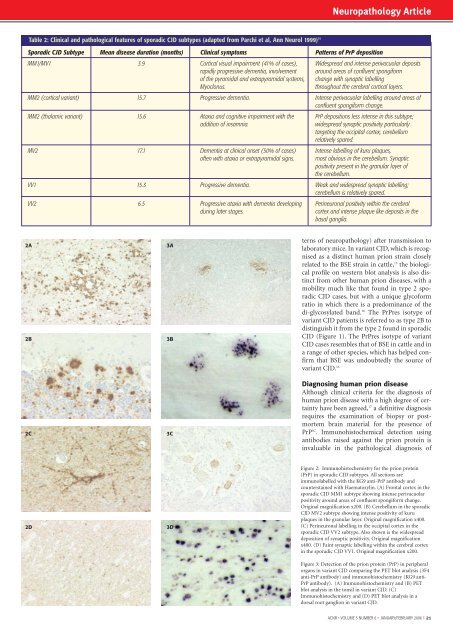
Neuropathology ArticleTable 2: Cl<strong>in</strong>ical <strong>and</strong> pathological features of sporadic CJD subtypes (adapted from Parchi et al, Ann Neurol 1999) 14Sporadic CJD Subtype Mean disease duration (months) Cl<strong>in</strong>ical symptoms Patterns of PrP depositionMM1/MV1 3.9 Cortical visual impairment (41% of cases), Widespread <strong>and</strong> <strong>in</strong>tense perivacuolar depositsrapidly progressive dementia, <strong>in</strong>volvement around areas of confluent spongiformof the pyramidal <strong>and</strong> extrapyramidal systems, change with synaptic labell<strong>in</strong>gMyoclonus.throughout the cerebral cortical layers.MM2 (cortical variant) 15.7 Progressive dementia. Intense perivacuolar labell<strong>in</strong>g around areas ofconfluent spongiform change.MM2 (thalamic variant) 15.6 Ataxia <strong>and</strong> cognitive impairment with the PrP depositions less <strong>in</strong>tense <strong>in</strong> this subtype;addition of <strong>in</strong>somnia.widespread synaptic positivity particularlytarget<strong>in</strong>g the occipital cortex; cerebellumrelatively spared.MV2 17.1 Dementia at cl<strong>in</strong>ical onset (50% of cases) Intense labell<strong>in</strong>g of kuru plaques,often with ataxia or extrapyramidal signs. most obvious <strong>in</strong> the cerebellum. Synapticpositivity present <strong>in</strong> the granular layer ofthe cerebellum.VV1 15.3 Progressive dementia. Weak <strong>and</strong> widespread synaptic labell<strong>in</strong>g;cerebellum is relatively spared.VV2 6.5 Progressive ataxia with dementia develop<strong>in</strong>g Per<strong>in</strong>euronal positivity with<strong>in</strong> the cerebraldur<strong>in</strong>g later stages.cortex <strong>and</strong> <strong>in</strong>tense plaque like deposits <strong>in</strong> thebasal ganglia.2A2B2C3A3B3Cterns of neuropathology) after transmission tolaboratory mice. In variant CJD, which is recognisedas a dist<strong>in</strong>ct human prion stra<strong>in</strong> closelyrelated to the BSE stra<strong>in</strong> <strong>in</strong> cattle, 15 the biologicalprofile on western blot analysis is also dist<strong>in</strong>ctfrom other human prion diseases, with amobility much like that found <strong>in</strong> type 2 sporadicCJD cases, but with a unique glycoformratio <strong>in</strong> which there is a predom<strong>in</strong>ance of thedi-glycosylated b<strong>and</strong>. 16 The PrPres isotype ofvariant CJD patients is referred to as type 2B todist<strong>in</strong>guish it from the type 2 found <strong>in</strong> sporadicCJD (Figure 1). The PrPres isotype of variantCJD cases resembles that of BSE <strong>in</strong> cattle <strong>and</strong> <strong>in</strong>a range of other species, which has helped confirmthat BSE was undoubtedly the source ofvariant CJD. 16Diagnos<strong>in</strong>g human prion diseaseAlthough cl<strong>in</strong>ical criteria for the diagnosis ofhuman prion disease with a high degree of certa<strong>in</strong>tyhave been agreed, 17 a def<strong>in</strong>itive diagnosisrequires the exam<strong>in</strong>ation of biopsy or postmortembra<strong>in</strong> material for the presence ofPrP SC . Immunohistochemical detection us<strong>in</strong>gantibodies raised aga<strong>in</strong>st the prion prote<strong>in</strong> is<strong>in</strong>valuable <strong>in</strong> the pathological diagnosis of2D3DFigure 2: Immunohistochemistry for the prion prote<strong>in</strong>(PrP) <strong>in</strong> sporadic CJD subtypes. All sections areimmunolabelled with the KG9 anti-PrP antibody <strong>and</strong>countersta<strong>in</strong>ed with Haematoxyl<strong>in</strong>. (A) Frontal cortex <strong>in</strong> thesporadic CJD MM1 subtype show<strong>in</strong>g <strong>in</strong>tense perivacuolarpositivity around areas of confluent spongiform change.Orig<strong>in</strong>al magnification x200. (B) Cerebellum <strong>in</strong> the sporadicCJD MV2 subtype show<strong>in</strong>g <strong>in</strong>tense positivity of kuruplaques <strong>in</strong> the granular layer. Orig<strong>in</strong>al magnification x400.(C) Per<strong>in</strong>euronal labell<strong>in</strong>g <strong>in</strong> the occipital cortex <strong>in</strong> thesporadic CJD VV2 subtype. Also shown is the widespreaddeposition of synaptic positivity. Orig<strong>in</strong>al magnificationx400. (D) Fa<strong>in</strong>t synaptic labell<strong>in</strong>g with<strong>in</strong> the cerebral cortex<strong>in</strong> the sporadic CJD VV1. Orig<strong>in</strong>al magnification x200.Figure 3: Detection of the prion prote<strong>in</strong> (PrP) <strong>in</strong> peripheralorgans <strong>in</strong> variant CJD compar<strong>in</strong>g the PET blot analysis (3F4anti-PrP antibody) <strong>and</strong> immunohistochemistry (KG9 anti-PrP antibody). (A) Immunohistochemistry <strong>and</strong> (B) PETblot analysis <strong>in</strong> the tonsil <strong>in</strong> variant CJD. (C)Immunohistochemistry <strong>and</strong> (D) PET blot analysis <strong>in</strong> adorsal root ganglion <strong>in</strong> variant CJD.<strong>ACNR</strong> • VOLUME 5 NUMBER 6 • JANUARY/FEBRUARY 2006 I 21