

PTI-609: A Novel Analgesic that Binds Filamin A to Control Opioid ...
PTI-609: A Novel Analgesic that Binds Filamin A to Control Opioid ...
PTI-609: A Novel Analgesic that Binds Filamin A to Control Opioid ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
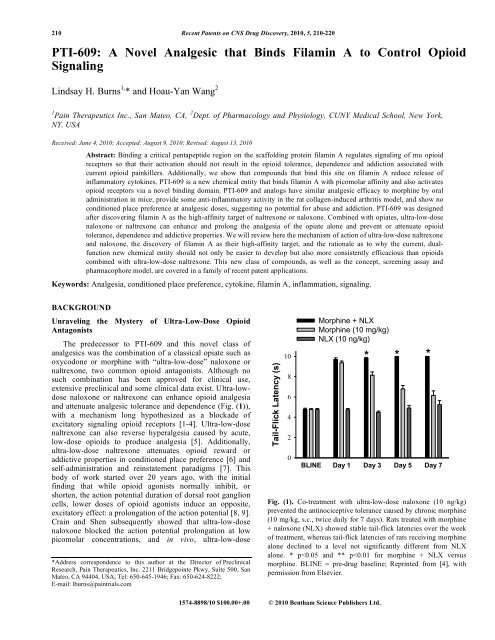
210 Recent Patents on CNS Drug Discovery, 2010, 5, 210-220<strong>PTI</strong>-<strong>609</strong>: A <strong>Novel</strong> <strong>Analgesic</strong> <strong>that</strong> <strong>Binds</strong> <strong>Filamin</strong> A <strong>to</strong> <strong>Control</strong> <strong>Opioid</strong>SignalingLindsay H. Burns 1, * and Hoau-Yan Wang 21 Pain Therapeutics Inc., San Mateo, CA, 2 Dept. of Pharmacology and Physiology, CUNY Medical School, New York,NY, USAReceived: June 4, 2010; Accepted: August 9, 2010; Revised: August 13, 2010Abstract: Binding a critical pentapeptide region on the scaffolding protein filamin A regulates signaling of mu opioidrecep<strong>to</strong>rs so <strong>that</strong> their activation should not result in the opioid <strong>to</strong>lerance, dependence and addiction associated withcurrent opioid painkillers. Additionally, we show <strong>that</strong> compounds <strong>that</strong> bind this site on filamin A reduce release ofinflamma<strong>to</strong>ry cy<strong>to</strong>kines. <strong>PTI</strong>-<strong>609</strong> is a new chemical entity <strong>that</strong> binds filamin A with picomolar affinity and also activatesopioid recep<strong>to</strong>rs via a novel binding domain. <strong>PTI</strong>-<strong>609</strong> and analogs have similar analgesic efficacy <strong>to</strong> morphine by oraladministration in mice, provide some anti-inflamma<strong>to</strong>ry activity in the rat collagen-induced arthritis model, and show noconditioned place preference at analgesic doses, suggesting no potential for abuse and addiction. <strong>PTI</strong>-<strong>609</strong> was designedafter discovering filamin A as the high-affinity target of naltrexone or naloxone. Combined with opiates, ultra-low-dosenaloxone or naltrexone can enhance and prolong the analgesia of the opiate alone and prevent or attenuate opioid<strong>to</strong>lerance, dependence and addictive properties. We will review here the mechanism of action of ultra-low-dose naltrexoneand naloxone, the discovery of filamin A as their high-affinity target, and the rationale as <strong>to</strong> why the current, dualfunctionnew chemical entity should not only be easier <strong>to</strong> develop but also more consistently efficacious than opioidscombined with ultra-low-dose naltrexone. This new class of compounds, as well as the concept, screening assay andpharmacophore model, are covered in a family of recent patent applications.Keywords: Analgesia, conditioned place preference, cy<strong>to</strong>kine, filamin A, inflammation, signaling.BACKGROUNDUnraveling the Mystery of Ultra-Low-Dose <strong>Opioid</strong>AntagonistsThe predecessor <strong>to</strong> <strong>PTI</strong>-<strong>609</strong> and this novel class ofanalgesics was the combination of a classical opiate such asoxycodone or morphine with “ultra-low-dose” naloxone ornaltrexone, two common opioid antagonists. Although nosuch combination has been approved for clinical use,extensive preclinical and some clinical data exist. Ultra-lowdosenaloxone or naltrexone can enhance opioid analgesiaand attenuate analgesic <strong>to</strong>lerance and dependence (Fig. (1)),with a mechanism long hypothesized as a blockade ofexcita<strong>to</strong>ry signaling opioid recep<strong>to</strong>rs [1-4]. Ultra-low-dosenaltrexone can also reverse hyperalgesia caused by acute,low-dose opioids <strong>to</strong> produce analgesia [5]. Additionally,ultra-low-dose naltrexone attenuates opioid reward oraddictive properties in conditioned place preference [6] andself-administration and reinstatement paradigms [7]. Thisbody of work started over 20 years ago, with the initialfinding <strong>that</strong> while opioid agonists normally inhibit, orshorten, the action potential duration of dorsal root ganglioncells, lower doses of opioid agonists induce an opposite,excita<strong>to</strong>ry effect: a prolongation of the action potential [8, 9].Crain and Shen subsequently showed <strong>that</strong> ultra-low-dosenaloxone blocked the action potential prolongation at lowpicomolar concentrations, and in vivo, ultra-low-dose*Address correspondence <strong>to</strong> this author at the Direc<strong>to</strong>r of PreclinicalResearch, Pain Therapeutics, Inc. 2211 Bridgepointe Pkwy, Suite 500, SanMateo, CA 94404, USA; Tel: 650-645-1946; Fax: 650-624-8222;E-mail: lburns@paintrials.comTail-Flick Latency (s)1086420Morphine + NLXMorphine (10 mg/kg)NLX (10 ng/kg)* * *BLINE Day 1 Day 3 Day 5 Day 7Fig. (1). Co-treatment with ultra-low-dose naloxone (10 ng/kg)prevented the antinociceptive <strong>to</strong>lerance caused by chronic morphine(10 mg/kg, s.c., twice daily for 7 days). Rats treated with morphine+ naloxone (NLX) showed stable tail-flick latencies over the weekof treatment, whereas tail-flick latencies of rats receiving morphinealone declined <strong>to</strong> a level not significantly different from NLXalone. * p
<strong>PTI</strong>-<strong>609</strong>, <strong>Novel</strong> <strong>Analgesic</strong> Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 211naltrexone (10ng/kg, s.c.) enhanced and prolonged morphine’santinociceptive potency [2]. This seminal report alsoincluded data demonstrating <strong>that</strong> co-treatment with ultralow-dosenaltrexone dramatically attenuated morphinewithdrawal and partially prevented analgesic <strong>to</strong>lerance.We demonstrated <strong>that</strong> the excita<strong>to</strong>ry signaling of opioidrecep<strong>to</strong>rs was due <strong>to</strong> their coupling <strong>to</strong> Gs proteins as earlierhypothesized, with one important distinction. Ultra-low-doseopioid antagonists were initially thought <strong>to</strong> bindpreferentially a subset of mu opioid recep<strong>to</strong>rs (MOR) [1, 10]<strong>that</strong> couple <strong>to</strong> Gs instead of the typical coupling <strong>to</strong> Gi andGo proteins <strong>that</strong> subsequently inhibit adenylyl cyclase [11].(The activated Gs proteins would instead stimulate adenylylcyclase and downstream cellular activities.) Our datasuggested <strong>that</strong> instead of binding a discreet population ofrecep<strong>to</strong>rs, ultra-low-dose naloxone or naltrexone controlledthe coupling preference of the MOR. Specifically, weshowed <strong>that</strong> co-treatment with 10ng/kg naloxone prevented achronic morphine-induced, Gi/o-<strong>to</strong>-Gs switch in G proteincoupling by the MOR as well as a coincident interaction ofthe G dimer with adenylyl cyclase II and IV [4]. Chronicmorphine treatment both diminished the native coupling <strong>to</strong>Gi/o and instated a novel coupling <strong>to</strong> Gs proteins by MOR.This chronic morphine induced MOR-Gs coupling was alsodemonstrated [4, 10] by researchers who initially postulated<strong>that</strong> G from Gi/o proteins was responsible for the excita<strong>to</strong>ryeffects of opiates [12]. We later confirmed <strong>that</strong> the G interacting with adenylyl cyclases originates from the Gsprotein coupling <strong>to</strong> MOR and not from MOR’s native Gproteins [13].The molecular pharmacology underlying ultra-low-dosenaloxone’s or naltrexone’s prevention of opioid <strong>to</strong>leranceand dependence was elucidated 10 years after the initialdemonstration of in vivo effects. It was clear <strong>that</strong> ultra-lowdosenaloxone blocked the MOR-Gs coupling associatedwith chronic opioid administration, while simultaneouslyenhancing levels of coupling <strong>to</strong> MOR’s native G proteins.Strikingly, our co-immunoprecipitation data showed <strong>that</strong> inspinal cord of rats cotreated with morphine plus ultra-lowdosenaloxone, MOR-Gi/o coupling levels greatly surpassedthose of opioid-naïve rats [4], perhaps mediating the enhancedanalgesia seen acutely with these combinations. Despitethis elucidation of mechanism of action of ultra-low-dosenaloxone or naltrexone, their actual binding site in regulatingMOR coupling was unknown.Finding the TargetBecause naloxone prevents MOR-Gs coupling atconcentrations well below its affinity for MOR and byinfluencing the coupling behavior of MORs, we consideredproteins <strong>that</strong> interact with MOR and MOR-associated Gproteins as the most likely targets, particularly those able <strong>to</strong>interact with multiple MORs. We first examined proteins<strong>that</strong> co-immunoprecipitated with Go-coupled MOR followingmorphine activation. We identified a 300kDa protein coimmunoprecipitatingwith Go-coupled MOR as filamin A(FLNA, 3CNK.pdb) and then demonstrated <strong>that</strong> naloxoneand naltrexone bind <strong>to</strong> FLNA with 4 pM affinity [14]. Thishigh-affinity interaction certainly could mediate the effectsof ultra-low doses of these two compounds.Best known for cross-linking cy<strong>to</strong>plasmic actin in<strong>to</strong>dynamic scaffolds <strong>to</strong> control cell motility, filamins are largecy<strong>to</strong>plasmic proteins increasingly found <strong>to</strong> regulate cellsignaling by interacting with over 30 different recep<strong>to</strong>rs andsignaling molecules [15, 16], including MOR [17]. Wededuced the precise, pentapeptide binding site on FLNA byusing overlapping peptides within the C-terminal, since C-terminal FLNA was earlier shown <strong>to</strong> interact with MORusing a yeast-two hybrid [17]. We demonstrated the functionalsignificance of this high-affinity interaction by usingpeptide fragments containing the binding site <strong>to</strong> preventnaloxone from binding full-length FLNA in organotypicstriatal slice cultures. Without addition of the peptide, FLNAinteracts with ultra-low-dose naloxone and naltrexone <strong>to</strong>prevent chronic morphine-induced MOR-Gs coupling, but inthe presence of the peptide fragment, naloxone loses itsability <strong>to</strong> block the Gs coupling (Fig. (2)). We also showed<strong>that</strong> this MOR-Gs coupling happens acutely but transiently,perhaps mediating the rewarding or euphoric properties ofopioids [18]. In <strong>that</strong> study, the cAMP second messenger wasmeasured and followed the timecourse of the Gs coupling(Fig. (3)). Co-treatment with naloxone, naltrexone or thepentapeptide FLNA fragment all blocked the increasedcAMP accumulation.This high-affinity binding site in C-terminal FLNAtherefore appears <strong>to</strong> underlie the paradoxical enhancement ofopioid analgesia and prevention of analgesic <strong>to</strong>lerance anddependence by ultra-low-dose opioid antagonists. The highaffinityinteraction of naloxone or naltrexone with FLNAlikely prevents a critical MOR-FLNA interaction <strong>that</strong>otherwise increases the probability of MOR coupling <strong>to</strong> Gsinstead of Gi/o proteins. In identifying the binding sitethough which ultra-low-dose naloxone or naltrexone preventsMOR-Gs coupling, our data also revealed an importantregulation of MOR-G protein coupling by filamin A.Additionally, the discovery of a novel, non-opioid bindingsite of naloxone and naltrexone underlying their “ultra-lowdose”effects opens avenues <strong>to</strong> developing a novel class ofanalgesics without the difficulties and constraints of combiningultra-low-dose opioid antagonists with opioidagonists.Variables in Defining an “Ultra-Low” DoseWhy was ultra-low-dose naltrexone or naloxone notconsistently effective? The most significant variable, somewhatsurprisingly, was the precise, ultra-low, dose. Ultralow-dosenaloxone and naltrexone effects do not follow atypical dose-response pattern, but usually span several logunits in the pg/kg <strong>to</strong> ng/kg range. There is a clear threshold,however, where increasing dose of naltrexone or naloxonestarts <strong>to</strong> disrupt their effects at ultra-low doses. Unfortunatelythis fine line seems <strong>to</strong> change with sex, strain, age,nociceptive versus neuropathic pain, and possibly manyother variables.An examination of the antinociceptive effects of variousratios of naltrexone:oxycodone (1:10 9 , 1:10 7 , and 1:10 5 )combined with a range of oxycodone doses (0.03-3mg/kg) inmale Swiss Webster mice illustrates the wide range ofeffective doses of naltrexone and the threshold whereanalgesic enhancement diminishes [19] Fig. (4). Whereas all
212 Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 Burns and WangFig. (2). FLNA 2561-2565 blocks 10 pM naloxone’s prevention of the chronic morphine-induced Go-<strong>to</strong>-Gs coupling switch. Striatal slices werechronically treated with vehicle, morphine, naloxone (NLX), morphine + NLX, or with one of the three FLNA peptides alone or incombination with morphine + NLX. Coupling between MOR and Gs/Go proteins was assessed by Western blot and analyzed bydensi<strong>to</strong>metric scanning. Chronic morphine exposure caused a Go-<strong>to</strong>-Gs coupling switch <strong>that</strong> was blocked by NLX co-treatment. NLX’ssuppression of this coupling switch was blocked by FLNA 2561-2570 but not by FLNA 2566-2575 or FLNA 2550-2560 , illustrating <strong>that</strong> NLX’sprotective effect occurs via its binding <strong>to</strong> FLNA within FLNA 2561-2570 . Solid bars indicate basal coupling; hatched bars indicate coupling afterin vitro morphine stimulation. n = 6. *p
<strong>PTI</strong>-<strong>609</strong>, <strong>Novel</strong> <strong>Analgesic</strong> Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 213Fig. (4). Dose response of ultra-low-dose naltrexone:oxycodone ratios in enhancing oxycodone antinociception in the 52ºC hot waterimmersion tail-flick tests in male Swiss Webster mice. Three different naltrexone:oxycodone ratios (1:10 9 , 1:10 7 , and 1:10 5 ) weresuperimposed on<strong>to</strong> a dose response of oxycodone (0.03, 0.1, 0.3, 1 and 3 mg/kg, s.c.). These naltrexone:oxycodone ratios enhanced theeffects of all doses of oxycodone, although the 1:10 9 and 1:10 7 ratios more potently enhanced the antinociceptive effect of the 3 mg/kgoxycodone dose than did the 1:10 5 dose ratio. Data are means ± s.e.m., n=3. Reprinted from [19] with permission from Research Signpost.The prevention of <strong>to</strong>lerance and withdrawal-associatedhyperalgesia by ultra-low-dose naltrexone was demonstratedat 0.3, 3, 300 and 3000ng/kg naltrexone when combined with3mg/kg morphine [20] and at 1pg/kg or 1ng/kg naltrexonewhen combined with oxycodone [21]. The higher naltrexonedoses used in these studies were used in males, suggestingmales are less sensitive <strong>to</strong> ultra-low-dose naltrexone effects.Hence, although male rodents are generally more sensitive <strong>to</strong>opioid analgesia [22], they appear <strong>to</strong> be less sensitive <strong>to</strong>ultra-low-dose naltrexone. These findings likely contribute<strong>to</strong> the variations in optimal ultra-low-dose naltrexone doseranges noted both with sex of the animal and the opioid usedin combination.In addition <strong>to</strong> sex differences, the effects of ultra-lowdosenaltrexone on morphine antinociception and <strong>to</strong>lerancecan also vary with rat strain [23]. Whereas the enhancemen<strong>to</strong>f morphine antinociception and attenuation of morphine<strong>to</strong>lerance was observed in both Sprague-Dawley and Long-Evans rats, naltrexone (0.1-100ng/kg) did not enhancemorphine antinociception in Fisher 344 or Lewis rats, nordid 10 or 100ng/kg naltrexone significantly attenuate<strong>to</strong>lerance in these two strains. Given <strong>that</strong> pg/kg doses ofnaltrexone most effectively enhanced oxycodone antinociceptionin female mice [21], perhaps this even lower doserange might enhance antinociception in these two rat strains.Terner (2006) also reported strain-dependent sex differences.Another example of strain differences is the initial report <strong>that</strong>ultra-low-dose naltrexone enhances morphine antinociceptionin Swiss Webster mice but antagonizes it in129/SvEv mice [24]. A final study in rats noted sex effects aswell as age effects: morphine antinociception was enhancedby low-dose naltrexone in mature female but not maturemale rats (18-22 weeks) and was negligible in younger rats[25]. However, the naltrexone dose range used in <strong>that</strong> studywas again comparatively high (0.002-2μg/kg), and the effectin mature females was “inversely related <strong>to</strong> dose.”Neuropathic pain is typically not well treated by opioids,but ultra-low-dose naltrexone can enhance their effects.Interestingly, the doses of naltrexone used <strong>to</strong> enhance theanti-hyperalgesic effects of oxycodone are notably higherthan the dose ranges mentioned above for nociceptive pain.In the spinal nerve ligation model of neuropathic pain,naltrexone enhanced the effectiveness of oxycodone andgreatly diminished the <strong>to</strong>lerance seen with repeated administrations[26]. Naltrexone was administered at 3μg/kg, a dose10-<strong>to</strong> 100-fold higher than those used <strong>to</strong> enhance analgesiaand alleviate <strong>to</strong>lerance in nociceptive pain.In summary, all of these studies show <strong>that</strong> an effectiveultra-low dose of naloxone or naltrexone seems <strong>to</strong> depend onmany interacting variables, including sex, strain, age, theopioid agonist used in combination, and type of pain. Thesevariables may reflect underlying differences in FLNAassociations, conformations or content.Clinical Experience with Ultra-Low-Dose <strong>Opioid</strong>AntagonistsThe inconsistent effects seen in clinical studies may alsobe related <strong>to</strong> the actual dose of naltrexone or naloxone andthe variables defining the <strong>to</strong>o-high threshold dose. Clinicalexperience with opioid antagonists combined with opiates islimited <strong>to</strong> small clinical studies [27-31], and two clinical
214 Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 Burns and Wangtrials [32, 33]. In a notable case study, a diabetic polyneuropathypatient, who previously had no pain relief from avariety of treatments, reported profound analgesia when2μg/day of naltrexone was co-administered with methadone[28]. The first controlled clinical study demonstrated anopioid-sparing effect and a reduction in side effects by acontinuous infusion of naloxone at 0.25μg/kg/hr added <strong>to</strong>morphine administered by patient controlled analgesia(PCA) [27]. Using nalmefene, an opioid antagonist not yetconfirmed <strong>to</strong> bind filamin A, another study reporteddecreased severity of pain 24 hours later and a decreasedneed for antiemetics and antipruretics following a single 15or 25μg injection prior <strong>to</strong> morphine PCA [31]. Cepeda andcolleagues were unable <strong>to</strong> replicate these effects using ahigher dose of naloxone mixed with morphine for PCA [29]and subsequently showed only a decrease in side effectsusing a much lower naloxone dose [30]. More recently,buprenorphine analgesia was significantly enhanced bynaloxone at 2.3μg for a 70kg patient but not by higher andlower doses of naloxone [34].Although it is difficult <strong>to</strong> compare doses of naloxone ornaltrexone in these small clinical studies, it is clear <strong>that</strong> theanalgesic-enhancing effect is not consistently seen, even ifdose levels seem <strong>to</strong> be consistent. A clear naltrexone doseeffectrelationship was noted, however, in two clinical trialsof Oxytrex TM (oxycodone + ultra-low-dose naltrexone). Thefirst showed a significant increase in analgesia over oxycodonealone if patients received 2 but not 4μg/day ofnaltrexone [32]. This study also showed a slight sex difference(Fig. (5)). In a phase III trial of Oxytrex, significantdecreases in physical dependence as well as in constipation,somnolence and pruritis were observed in patients receiving2 but not 4μg/day of naltrexone [33]. In this latter trial,patients were allowed <strong>to</strong> titrate their dose by taking higher(or lower) strength tablets <strong>to</strong> achieve sufficient analgesia, butwith the naltrexone in Oxytrex tablets fixed at 1μg/tablet,patients received a consistent 2 or 4μg naltrexone/day,depending on bid or qid dose regimens. Interestingly, the<strong>to</strong>tal average daily dose after self-titration was 12% lowerfor both bid and qid Oxytrex groups compared <strong>to</strong> the qidgroup receiving oxycodone with no naltrexone.Two Binding Sites on <strong>Filamin</strong> AOne wonders how it can be <strong>that</strong> 2 but not 4μg/day ofnaltrexone can enhance analgesia clinically, and how such abroad dose range below this threshold can be effective whilecrossing the line, as variable as it is, destroys the effects.Intuitively, the obvious thought was <strong>that</strong> the additionalopioid antagonist starts <strong>to</strong> disrupt the opioid agonist, therebydiminishing the overall effect. However, a nanogram ormicrogram dose of opioid antagonist is not likely <strong>to</strong>noticeably disrupt the biological effects of a milligram doseof opioid agonist. Evidence <strong>that</strong> the possibility of spillover <strong>to</strong>opioid recep<strong>to</strong>rs is not the case comes from a study examiningthe reduction in rewarding properties of oxycodoneby ultra-low-dose naltrexone [6]. In this study, 0.03 or0.3ng/kg naltrexone blocked a conditioned place preference(CPP) <strong>to</strong> 3mg/kg oxycodone, whereas 3ng/kg naltrexone didnot block this CPP <strong>to</strong> oxycodone (Fig. (6)). As thenaltrexone dose increased <strong>to</strong> 30ng/kg, the CPP <strong>to</strong> oxycodoneis not significant. The robust CPP in the middle of this doseresponsecurve demonstrates a point where the “ultra-lowdoseeffects” are lost and the classical antagonism of opioidrecep<strong>to</strong>rs is not yet occurring. In analgesic paradigms, ifopioid recep<strong>to</strong>rs are antagonized, analgesia would bedecreased, just as it would when crossing the ultra-low-dosethreshold. However, in the CPP paradigm, when the ultra-Fig. (5). Reduction in pain intensity in males and females by Oxytrex. Oxytrex BID, a daily dose of 2 μg naltrexone, provided the greatestreduction in pain intensity scores in both males and females. At Week 3, Oxytrex BID was significantly better than placebo in males andsignificantly better than oxycodone in females. The pain intensity scores of the Oxytrex QID group, receiving 4 μg naltrexone/day, did notstatistically separate from placebo. Reprinted from [32] with permission from Elsevier.
<strong>PTI</strong>-<strong>609</strong>, <strong>Novel</strong> <strong>Analgesic</strong> Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 215Fig. (6). Dose-response of naltrexone on a CPP <strong>to</strong> oxycodone. Bars represent the mean (± SEM) amount of time (s) spent in saline- and drugpairedcompartments on test day. Oxycodone (3mg/kg, s.c.) produced a significant CPP <strong>that</strong> was blocked by the addition of naltrexone(NTX) at 0.03ng/kg s.c., or 0.3ng/kg. In contrast, NTX at 3ng/kg s.c. did not block the CPP <strong>to</strong> oxycodone. Oxycodone combined with thehighest dose of NTX, 30ng/kg s.c., produced only a trend <strong>to</strong>ward a CPP. Reprinted from [6] with permission from Springer-Verlag.low-dose effect is lost, the CPP <strong>to</strong> oxycodone reappears;whereas, antagonizing opioid recep<strong>to</strong>rs would interfere withthe CPP.What then is disrupting the ultra-low-dose naloxone/naltrexone effects, making this threshold of efficacy such afine line <strong>to</strong> tread? We believe this threshold is due <strong>to</strong> asecond binding site of naloxone and naltrexone on the FLNAprotein. A competition (displacement) curve for the inhibitionof tritiated naloxone binding by naltrexone <strong>to</strong>membranes from FLNA-expressing cells shows two affinitystates with IC 50 -high of 3.94 picomolar and IC 50 -low of 834picomolar Fig. (7) [14]. It is very possible <strong>that</strong> binding thealmost nanomolar binding site on FLNA may alter theconformation of the protein or disrupt some conformationalchange induced by binding the picomolar binding site. Thistwo-hundred-fold difference in affinity could represent thefine line of whether ultra-low-dose naloxone or naltrexoneenhances opioid analagesia, prevents <strong>to</strong>lerance and dependenceand attenuates opioid reward or not.A NEW APPROACH: <strong>PTI</strong>-<strong>609</strong>Now <strong>that</strong> the high-affinity binding site of naloxone andnaltrexone is known <strong>to</strong> be a pentapeptide region on FLNA,we were able <strong>to</strong> design novel compounds <strong>that</strong> combine highaffinityFLNA binding with MOR agonist activity in asingle, new chemical entity. Our approach was molecularmodeling, virtual screening of the world’s compoundsagainst the model, followed by actual screening of a smallnumber of selected compounds for binding <strong>to</strong> the FLNApentapeptide and activation of MOR. The screening data wasused <strong>to</strong> refine the model, and the cycle was repeated oncemore before initiating medicinal chemistry, which producedseveral good lead candidates with fM <strong>to</strong> pM affinity <strong>to</strong> theFLNA pentapeptide and good analgesic efficacy in mice byoral administration. The analgesic dose-response curve oforally administered <strong>PTI</strong>-<strong>609</strong> indicates <strong>that</strong> <strong>PTI</strong>-<strong>609</strong> isbioavailable and CNS penetrable Fig. (8). The initial patentapplication covered the concept, the screening assay and anearly pharmacophore model [35], and several patentapplications covering families of these novel compoundshave also published [36-38]. These novel compounds do notstructurally resemble known opioids, and interestingly, thethree lead compounds <strong>that</strong> were tested do not displacediprenorphine in a binding assay, suggesting a novel bindingdomain in activating MOR. Functional activation of MOR inhuman post-mortem caudate tissue using a GTPS bindingassay shows agonist activity comparable <strong>to</strong> the full agonist[D-Ala 2 , N-MePhe 4 , Gly-ol]-enkephalin (DAMGO). At 0.1μM concentration, stimulation of binding was 204.6 ± 52.6%for <strong>PTI</strong>-<strong>609</strong> and 210.6 ± 34.0% for DAMGO.The dual function of <strong>PTI</strong>-<strong>609</strong> avoids the difficulties ofdeveloping a combination product, allowing easier formulationand simpler clinical trials. Most importantly, byscreening for compounds <strong>that</strong> bind only the picomolarbinding site on FLNA and not the second, lower affinity siteon this scaffolding protein, we avoid the difficulties intreading the ever-moving, fine line of the ultra-low dose ofnaloxone or naltrexone. To confirm <strong>that</strong> this sand-trap is nowa non-issue, we can simply increase concentrations of thesecompounds and confirm <strong>that</strong> effects are not lost (see Antiinflamma<strong>to</strong>ryeffects section). What is expected is an analgesicsuited for moderate-<strong>to</strong>-severe pain with little <strong>to</strong> no
216 Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 Burns and WangFig. (7). Naloxone binds FLNA with picomolar affinity. A competition (displacement) curve for the inhibition of [ 3 H]-naloxone(NLX)binding by naltrexone <strong>to</strong> membranes from FLNA-expressing A7 cells shows two affinity states with IC 50 -H of 3.94 picomolar and IC 50 -L of834 picomolar. A nonlinear curve-fit analysis was performed using a competition equation <strong>that</strong> assumed two saturable sites for thenaltrexone curve comprising of 16 concentrations ranging from 0.1pM <strong>to</strong> 1μM. Data are derived from 6 experiments each using a differentset of A7 cells. Reprinted from [14].Fig. (8). <strong>Analgesic</strong> efficacy of <strong>PTI</strong>-<strong>609</strong> by oral administration in mice. Mice were tested in the 52 o C hot water immersion test using a 10-scu<strong>to</strong>ff, which represents 100% maximal possible effect (MPE). Data are means ± SEM. n=6.<strong>to</strong>lerance, dependence and addictive potential. Additionally,we anticipated some anti-inflamma<strong>to</strong>ry activity.Anti-Inflamma<strong>to</strong>ry EffectsWhy would we expect anti-inflamma<strong>to</strong>ry effects fromcompounds <strong>that</strong> bind this site on FLNA? The labora<strong>to</strong>ry ofJau-Shoyng Hong has shown <strong>that</strong> both (+) and (-)naloxoneinhibit lipopolysaccaride (LPS)-induced activation ofmicroglia and release of proinflamma<strong>to</strong>ry fac<strong>to</strong>rs in mixedneuron-glial cultures at fem<strong>to</strong>molar concentrations [39, 40].The authors describe this anti-inflamma<strong>to</strong>ry andneuroprotective response as bimodal, because the protectionis lost in the high picomolar range before reappearing in themicromolar range. This threshold is reminiscent of the lossof effects of naloxone and naltrexone in potentiating opioidanalgesia, and knowing <strong>that</strong> naltrexone binds FLNA in thefem<strong>to</strong>molar <strong>to</strong> picomolar range, we suspected <strong>that</strong> these
<strong>PTI</strong>-<strong>609</strong>, <strong>Novel</strong> <strong>Analgesic</strong> Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 217effects could be the result of FLNA binding. The Honglabora<strong>to</strong>ry suggested NADPH oxidase as a target, whichappears <strong>to</strong> be a critical final common pathway enzyme inproducing both extracellular and intracellular reactiveoxygen species, and subsequently, inflamma<strong>to</strong>ry cy<strong>to</strong>kinerelease.The Linda R. Watkins labora<strong>to</strong>ry has also demonstrated<strong>that</strong> naloxone and naltrexone block glial activation andresulting proinflamma<strong>to</strong>ry cy<strong>to</strong>kine release [41]. The targetsuggested by the Watkins labora<strong>to</strong>ry in these effects is <strong>to</strong>lllikerecep<strong>to</strong>r 4 (TLR4) of innate immune cells, becausenaloxone and naltrexone, including their isomers <strong>that</strong> areinactive at opioid recep<strong>to</strong>rs, block TLR4 signaling [41, 42].A large body of work, led by Dr. Watkins, has demonstratedthe role of glial activation in the negative effects of opioids,including opioid <strong>to</strong>lerance, dependence, reward, andrespira<strong>to</strong>ry depression [43], so it is possible <strong>that</strong> ultra-lowdoseopioid antagonists are alleviating these effects bydiminishing the glial activation of opioids. Although the glialactivation could be a downstream effect of MOR-Gssignaling, the cell line used <strong>to</strong> demonstrate <strong>that</strong> (+) and (-)naloxone disrupt TLR4 signaling was devoid of opioidrecep<strong>to</strong>rs [42]. Hence, binding <strong>to</strong> FLNA in glial cells mayslow down vesicular release of cy<strong>to</strong>kines, or even disruptTLR4 signaling. Although FLNA binders such as(+)naloxone have not been reported <strong>to</strong> have antinociceptiveeffects themselves, they can reduce allodynia in a model ofneuropathic pain [40], and this effect is attributed <strong>to</strong> theirsuppression of glial activation.We first assessed cy<strong>to</strong>kine release from LPS-stimulatedhuman astrocytes and the potential blockade of this cy<strong>to</strong>kinerelease by <strong>PTI</strong>-<strong>609</strong> and related novel compounds. Weassessed concentrations from 100 fM <strong>to</strong> 100 nM andcompared these <strong>to</strong> 10 pM and 1 nM of (+)naloxone. Whereas<strong>PTI</strong>-<strong>609</strong> and two related compounds potently reducedrelease of IL-6, IL-1 and TNF-, (+)naloxone suppressedrelease of these cy<strong>to</strong>kines somewhat less potently at 10 pMand virtually not at all at 1 nM (Wang and Burns, unpublisheddata, Fig. (9)). This loss of efficacy by (+)naloxone at1 nM again demonstrates the crossing of the ultra-low-dosethreshold, likely mediated by binding <strong>to</strong> the second bindingsite on FLNA, the site it binds with 834 pM affinity. Thelack of dose-response in this initial test of cy<strong>to</strong>kine releasesuggests <strong>that</strong> naloxone and naltrexone’s picomolar bindingsite is already saturated by these com-pounds at 100 fM.Lower concentrations may show a true dose-response.We next assessed anti-inflamma<strong>to</strong>ry activity of <strong>PTI</strong>-<strong>609</strong>in the rat collagen-induced arthritis model. Animals weredosed twice daily by oral gavage beginning on the day of thesecond collagen injection. Three different dose groups of thenovel compound were compared <strong>to</strong> 3mg/kg celecoxib and25mg/kg ibuprofen. <strong>PTI</strong>-<strong>609</strong> significantly reduced footvolume at 56mg/kg and at one time point at 5.6mg/kg, butnot at 32mg/kg. The reduction in foot volume was far lesspotent than <strong>that</strong> produced by celecoxib, and approximatelyhalf <strong>that</strong> seen with ibuprofen in this initial proof of conceptstudy (Wang and Burns, unpublished data, Fig. (10).Percent Decrease100806040200100 fM10 pM1 nM100 nMCpd 1 Cpd 2 <strong>PTI</strong>-<strong>609</strong> (+)NaloxoneFig. (9). FLNA-binding compounds block LPS-stimulated cy<strong>to</strong>kinerelease from human astrocytes. Three FLNA-binding compoundsblock TNF- release from primary cultures of human astrocytes by75% or more at concentrations ranging from 100 fM <strong>to</strong> 100 nM.(+)Naloxone blocked TNF- release by almost 60% at 10 pM, butthis suppression was nearly abolished at 1 nM. Similar results wereobtained with IL-1 and IL-6. Data are means ± SEM. n=4.Addictive PotentialOur data linking the immediate and transient MOR-Gscoupling <strong>to</strong> CREB activation, as well as the blockade ofconditioned place preference (CPP) by pg/kg doses ofnaltrexone, suggested <strong>that</strong> our novel compounds would havereduced addictive potential. We previously showed <strong>that</strong> inbrain slices, acute high dose morphine caused an immediatebut transient Gs coupling by MOR <strong>that</strong> was associated withactivation of cyclic AMP response element-binding protein(CREB) [18], a transcription fac<strong>to</strong>r implicated in addictionand withdrawal effects [44-48]. Blockade of this immediateMOR-Gs coupling by picomolar concentrations of naloxoneor by its pentapeptide binding site on FLNA blocked theassociated activation of CREB. The rewarding effects ofopioid drugs are most pronounced as plasma levels are risingquickly and are less intense or even negative later [49-51],suggesting <strong>that</strong> this immediate Gs coupling is at leasttemporally associated with the most intense phase of opioidreward. To support the hypothesis <strong>that</strong> our FLNA-bindingnovel compounds would not have the same rewardingproperties of classical opioids, we examined equi-analgesicdoses of <strong>PTI</strong>-<strong>609</strong> and oxycodone in the CPP paradigm. Oraldoses <strong>that</strong> produced an 80-90% analgesic effect as well as adose log lower were compared <strong>to</strong> a vehicle group, usingthree conditioning sessions <strong>to</strong> the drug-paired compartmentas well as <strong>to</strong> the vehicle-paired compartment. A significantCPP <strong>to</strong> both doses of oxycodone was observed, whereas incontrast, there was no change from baseline in time spent inthe putative conditioning side with either dose of <strong>PTI</strong>-<strong>609</strong>
218 Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 Burns and WangFig. (10). Reduction in foot swelling in rat collagen-induced arthritis model. Oral twice daily administration of <strong>PTI</strong>-<strong>609</strong> reduced footswelling in female Wistar rats immunized with type-II collagen on days 1 and 9. <strong>PTI</strong>-<strong>609</strong> was administered at 5.6, 32 and 56 mg/kg startingon day 9. Significant differences from vehicle (p
<strong>PTI</strong>-<strong>609</strong>, <strong>Novel</strong> <strong>Analgesic</strong> Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 219xone, <strong>PTI</strong>-<strong>609</strong> avoids the second, lower-affinity binding siteon FLNA and therefore should maintain its beneficial effectswith increasing dose, allowing the picomolar site <strong>to</strong> saturatewith no interference. Although much work remains before<strong>PTI</strong>-<strong>609</strong> can enter clinical trials, the early research presentedhere shows <strong>PTI</strong>-<strong>609</strong> and related compounds <strong>to</strong> be efficaciousanalgesics <strong>that</strong> possess additional anti-inflamma<strong>to</strong>ry propertiesand, according <strong>to</strong> the standard conditioned placepreference paradigm, may not carry the abuse and addictivepotential of classical opioid drugs. The problems withcurrent opioid painkillers, from side effects <strong>to</strong> <strong>to</strong>lerance,dependence, abuse, diversion and addiction demonstrate agreat need for a better class of efficacious analgesics. <strong>PTI</strong>-<strong>609</strong> is a promising new drug candidate <strong>that</strong> seems wellpoised <strong>to</strong> fulfill <strong>that</strong> need.ACKNOWLEGEMENTSPain Therapeutics, Inc. acknowledges the followingcontracted services: pharmacophore modeling and in silicoscreening (Jian Wang); analgesia and conditioned placepreference testing (Ed Bilsky of AIV Testing, Biddeford,ME); medicinal chemistry, synthesis and CIA model (Medicilon,Inc., Shanghai, China). All in vitro screening for leadselection was performed by H.-Y. Wang at CUNY MedicalSchool under a research collaboration.CONFLICT OF INTERESTL.H. Burns is an employee of Pain Therapeutics Inc., thecompany <strong>that</strong> funded the development work on <strong>PTI</strong>-<strong>609</strong> aswell as earlier work on Oxytrex.REFERENCES[1] Crain SM, Shen K-F. Antagonists of excita<strong>to</strong>ry opioid recep<strong>to</strong>rfunctions enhance morphine's analgesic potency and attenuateopioid <strong>to</strong>lerance/dependence liability. Pain 2000; 84: 121-31.[2] Crain SM, Shen K-F. Ultra-low concentrations of naloxoneselectively antagonize excita<strong>to</strong>ry effects of morphine on sensoryneurons, thereby increasing its antinociceptive potency andattenuating <strong>to</strong>lerance/dependence during chronic cotreatment. ProcNatl Acad Sci USA 1995; 92: 10540-4.[3] Powell KJ, Abul-Husn NS, Jhamandas A, Olmstead MC, BeningerRJ, Jhamandas K. Paradoxical effects of the opioid antagonistnaltrexone on morphine analgesia, <strong>to</strong>lerance, and reward in rats. JPharmacol Exp Ther 2002; 300: 588-96.[4] Wang H-Y, Friedman E, Olmstead MC, Burns LH. Ultra-low-dosenaloxone suppresses opioid <strong>to</strong>lerance, dependence and associatedchanges in Mu opioid recep<strong>to</strong>r-G protein coupling and Gsignaling. Neuroscience 2005; 135: 247-61.[5] Crain SM, Shen K-F. Acute thermal hyperalgesia elicited by lowdosemorphine in normal mice is blocked by ultra-low-dosenaltrexone, unmasking potent opioid analgesia. Brain Res 2001;888: 75-82.[6] Olmstead MC, Burns LH. Ultra-low-dose naltrexone suppressesrewarding effects of opiates and aversive effects of opiatewithdrawal in rats. Psychopharmacol 2005; 181: 576-81.[7] Leri F, Burns LH. Ultra-low-dose naltrexone reduces the rewardingpotency of oxycodone and relapse vulnerability in rats. PharmacolBiochem Behav 2005; 82: 252-62.[8] Shen KF, Crain SM. Dual opioid modulation of the action potentialduration of mouse dorsal root ganglion neurons in culture. BrainRes 1989; 491: 227-42.[9] Crain SM, Shen K-F. <strong>Opioid</strong>s can evoke direct recep<strong>to</strong>r-mediatedexcita<strong>to</strong>ry effects on sensory neurons. Trends Pharmacol Sci 1990;11: 77-81.[10] Chakrabarti S, Regec A, Gintzler AR. Biochemical demonstrationof mu-opioid recep<strong>to</strong>r association with Gs: Enhancementfollowing morphine exposure. Mol Brain Res 2005; 135: 217-24.[11] Laugwitz KL, Offermanns S, Spicher K, Schultz G. Mu and deltaopioid recep<strong>to</strong>rs differentially couple <strong>to</strong> G protein subtypes inmembranes of human neuroblas<strong>to</strong>ma SH-SY5Y cells. Neuron 1993;10: 233-42.[12] Gintzler AR, Chakrabarti S. <strong>Opioid</strong> <strong>to</strong>lerance and the emergence ofnew opioid recep<strong>to</strong>r-coupled signaling. Mol Neurobiol 2001; 21:21-33.[13] Wang H-Y, Burns LH. G <strong>that</strong> interacts with adenylyl cyclase inopioid <strong>to</strong>lerance originates from a Gs protein. J Neurbiol 2006; 66:1302-10.[14] Wang H-Y, Frankfurt M, Burns LH. High-affinity naloxone binding<strong>to</strong> filamin A prevents mu opioid recep<strong>to</strong>r - Gs coupling underlyingopioid <strong>to</strong>lerance and dependence. PLoS One 2008; 3(2): e1554.[15] Feng Y, Walsh C. The many faces of filamin: A versatile molecularscaffold for cell motility and signalling. Nat Cell Biol 2004; 6:1034-8.[16] S<strong>to</strong>ssel T, Condeelis J, Cooley L, Hartwig J, Noegel A, SchleicherM, et al. <strong>Filamin</strong>s as integra<strong>to</strong>rs of cell mechanics and signalling.Nature 2001; 2: 138-45.[17] Onoprishvili I, Andria M, Kramer H, Ancevska-Taneva N, Hiller J,Simon E. Interaction between the μ opioid recep<strong>to</strong>r and fliamin A isinvolved in recep<strong>to</strong>r regulation and trafficking. Mol Pharmacol2003; 64: 1092-100.[18] Burns LH. Ultra-low-dose opioid antagonists enhance opioidanalgesia while reducing <strong>to</strong>lerance, dependence and addictiveproperties. In: Capasso A, Ed. Recent Developments in PainResearch. Kerala, India: Research Signpost 2005; 115-36.[19] Shen K-F, Crain SM, Moate P, Bos<strong>to</strong>n R, de Kater AW,Schoenhard GL. <strong>PTI</strong>-555, reverses and prevents morphine-induced<strong>to</strong>lerance and naloxone-precipitated withdrawal in mice chronicallytreated with morphine. J Pain 2002; 3: 50.[20] Shen K-F, Crain SM, Moate P, Bos<strong>to</strong>n R, de Kater AW,Schoenhard GL. <strong>PTI</strong>-801, a novel formulation of oxycodone, showsabsence of <strong>to</strong>lerance, physical dependence and naloxoneprecipitatedwithdrawal effects in mice. J Pain 2002; 3: 49.[21] Kest B, Sar<strong>to</strong>n E, Dahan A. Gender differences in opioid-mediatedanalgesia. Anesthesiology 2000; 93: 539-47.[22] Terner JM, Barrett AC, Lomas LM, Negus SS, Picker MJ.Influence of low doses of naltrexone on morphine antinociceptionand morphine <strong>to</strong>lerance in male and female rats of four strains. Pain2006; 122: 90-101.[23] Crain SM, Shen K-F. Enhanced analgesic potency and reduced<strong>to</strong>lerance of morphine in 129/SvEv mice: evidence for a deficiencyin GM1 ganglioside-regulated excita<strong>to</strong>ry opioid recep<strong>to</strong>r functions.Brain Res 2000; 856: 227-35.[24] Hamman SR, Malik H, Sloan JW, Wala EP. Interactions of "ultralow"doses of naltrexone and morphine in mature and young maleand female rats. Recept Chan 2004; 10: 73-81.[25] Largent-Milnes TM, Guo W, Wang H-Y, Burns LH, Vanderah T.Oxycodone plus ultra-low-dose naltrexone attenuates neuropathicpain and associated μ opioid recep<strong>to</strong>r - Gs coupling. J Pain 2008; 9:700-13.[26] Gan TJ, Ginsberg B, Glass PSA, Fortney J, Jhaveri R, Perno R.<strong>Opioid</strong>-sparing effects of a low-dose infusion of naloxone inpatient-administered morphine sulfate. Anesthesiology 1997; 87:1075-81.[27] Cruciani RA, Lussier D, Miller-Saultz D, Arbuck DM. Ultra-lowdose oral naltrexone decreases side effects and potentiates the effec<strong>to</strong>f methadone. J Pain Symp<strong>to</strong>m Manage 2003; 25: 491-4.[28] Cepeda MS, Africano JM, Manrique AM, Fragoso W, Carr DB.The combination of low dose naloxone and morphine in PCA doesnot decrease opioid requirements in the pos<strong>to</strong>perative period. Pain2002; 96: 73-9.[29] Cepeda MS, Alvarez H, Morales O, Carr DB. Addition of ultralowdose naloxone <strong>to</strong> pos<strong>to</strong>perative morphine PCA: Unchangedanalgesia and opioid requirement but decreased incidence of opioidside effects. Pain 2004; 107: 41-6.[30] Joshi GP, Duffy L, Chehade J, Wesevich J, Gajraj N, Johnson ER.Effects of prophylactic nalmefene on the incidence of morphinerelatedside effects in patients receiving intravenous patientcontrolledanalgesia. Anesthesiology 1999; 90: 1007-11.
220 Recent Patents on CNS Drug Discovery, 2010, Vol. 5, No. 3 Burns and Wang[31] Chindalore VL, Craven RA, Butera PG, Yu KP, Burns LH,Friedmann N. Adding ultra-low-dose naltrexone <strong>to</strong> oxycodoneenhances and prolongs analgesia. J Pain 2005; 6: 392-9.[32] Webster LR, Butera PG, Moran LV, Wu N, Burns LH, FriedmannN. Oxytrex minimizes physical dependence while providing effectiveanalgesia: A randomized controlled trial in low-back pain. JPain 2006; 7(12): 937-46.[33] La Vincente S, White J, Somogyi A, Bochner F, Chapleo C.Enhanced buprenorphine analgesia with the addition of ultra-lowdosenaloxone in healthy subjects. Clin Pharmacol Ther 2008;83(1): 144-52.[34] Wang, H.-Y., Barbier, L., Wang, J. Analgesia with minimal<strong>to</strong>lerance and dependence by a mu opioid recep<strong>to</strong>r agonist <strong>that</strong> alsobinds filamin A. WO2009059225 (2009).[35] Barbier, L., Wang, H.-Y., Lin, N.-H., Blasko, A. <strong>Analgesic</strong> <strong>that</strong>binds filamin A. WO201051374 (2010).[36] Barbier, L., Wang, H.-Y., Lin, N.-H., Blasko, A. <strong>Filamin</strong> A-bindinganti-inflamma<strong>to</strong>ry analgesic. WO2010051476 (2010).[37] Barbier, L., Wang, H.-Y., Lin, N.-H, Blasko, A. <strong>Filamin</strong> A-bindinganti-inflamma<strong>to</strong>ry analgesic. WO2010051497 (2010).[38] Liu B, Du L, Hong J-S. Naloxone protects rat dopaminergicneurons against inflamma<strong>to</strong>ry damage through inhibition ofmicroglial activation and superoxide generation. J Pharmacol ExpTher 2000; 293: 607-617.[39] Qin L, Block M, Liu Y, Biens<strong>to</strong>ck R, Pei Z, Zhang W, et al.Microglial NADPH oxidase is a novel target for fem<strong>to</strong>molarneuroprotection against oxidative stress. FASEB 2005; 19: 550-7.[40] Hutchinson M, Zhang Y, Brown K, Coats B, Shridhar M, Sholar P,et al. Non-stereoselective reversal of neuropathic pain by naloxoneand naltrexone. Eur J Neurosci 2008; 28: 20-9.[41] Hutchinson M, Zhang Y, Shridhar M, Evans J, Buchanan M, ZhaoT. Evidence <strong>that</strong> opioids may have <strong>to</strong>ll-like recep<strong>to</strong>r 4 and MD-2effects. Brain Behav Immun 2010; 24: 83-95.[42] Hutchinson M, Bland S, Johnson K, Rice K, Maier S, Watkins LR.<strong>Opioid</strong>-induced glial activation: Mechanisms of activation andimplications for opioid analgesia, dependence and reward.ScientificWorld J 2007; 7: 98-111.[43] Wang H-Y, Burns L. Naloxone’s pentapeptide binding site onfilamin A blocks mu opioid recep<strong>to</strong>r–Gs coupling and CREBactivation of acute morphine. PLoS ONE 2009; 4: e4282.[44] Carlezon W, Duman R, Nestler EJ. The many faces of CREB.Trends Neurosci 2005; 28: 436-45.[45] Guitart X, Thompson M, Mirante C, Greenberg M, Nestler EJ.Regulation of cyclic AMP response element-binding protein(CREB) phosphorylation by acute and chronic morphine. JNeurochem 1992; 58: 1168-71.[46] Nestler EJ. Molecular neurobiology of addiction. Am J Addict2001; 10: 201-17.[47] Nestler EJ. Molecular mechanisms of drug addiction.Neuropharmacology 2004; 47: 24-32.[48] Shaw-Lutchman T, Barrot M, Wallace T, Glilden L, Zachariou V,Impey S, et al. Regional and cellular mapping of cAMP responseelement-mediated transcription during naltrexone-precipitatedmorphine withdrawal. J Neurosci 2002; 22: 3663-72.[49] Marsch L, Bickel W, Badger G, Rathmell J, Swedberg M, BrorJonzon B, et al. Effects of infusion rate of intravenouslyadministered morphine on physiological, psychomo<strong>to</strong>r, and selfreportedmeasures in humans. J Pharmacol Exp Ther 2001; 299:1056-65.[50] Jasinski D, Pevnick J, Griffith J. Human pharmacology and abusepotential of the analgesic buprenorphine. Arch Intern Psychiat1978; 35: 501-16.[51] Zacny J, Gutierrez S. Characterizing the subjective, psychomo<strong>to</strong>r,and physiological effects of oral oxycodone in non-drug-abusingvolunteers. Psychopharm 2003; 170: 242-54.