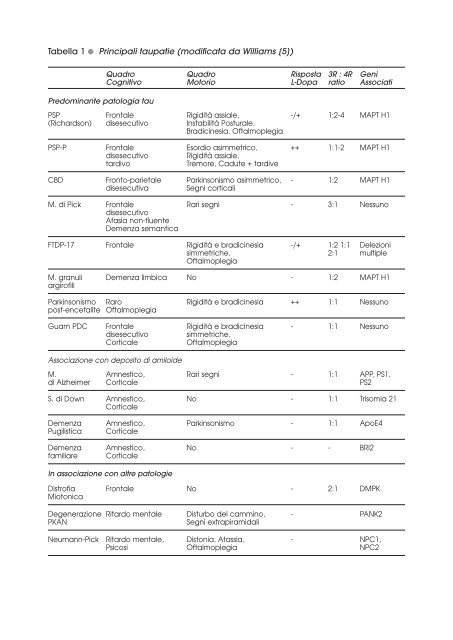
Tabella 1 ● Principali <strong>taupatie</strong> (modificata da Williams [5])Pr<strong>ed</strong>ominante patologia tauQuadro Quadro Risposta 3R : 4R GeniCognitivo Motorio L-Dopa ratio AssociatiPSP Fronta<strong>le</strong> Rigidità assia<strong>le</strong>, -/+ 1:2-4 MAPT H1(Richardson) disesecutivo Instabilità Postura<strong>le</strong>,Bradicinesia, Oftalmop<strong>le</strong>giaPSP-P Fronta<strong>le</strong> Esordio asimmetrico, ++ 1:1-2 MAPT H1disesecutivoRigidità assia<strong>le</strong>,tardivoTremore, Cadute + tardiveCBD Fronto-parieta<strong>le</strong> Parkinsonismo asimmetrico, - 1:2 MAPT H1disesecutivaSegni corticaliM. di Pick Fronta<strong>le</strong> Rari segni - 3:1 NessunodisesecutivoAfasia non-fluenteDemenza semanticaFTDP-17 Fronta<strong>le</strong> Rigidità e bradicinesia -/+ 1:2 1:1 De<strong>le</strong>zionisimmetriche, 2:1 multip<strong>le</strong>Oftalmop<strong>le</strong>giaM. granuli Demenza limbica No - 1:2 MAPT H1argirofiliParkinsonismo Raro Rigidità e bradicinesia ++ 1:1 Nessunopost-encefalite Oftalmop<strong>le</strong>giaGuam PDC Fronta<strong>le</strong> Rigidità e bradicinesia - 1:1 Nessunodisesecutivosimmetriche,Cortica<strong>le</strong>Oftalmop<strong>le</strong>giaAssociazione con deposito di amiloideM. Amnestico, Rari segni - 1:1 APP, PS1,di Alzheimer Cortica<strong>le</strong> PS2S. di Down Amnestico, No - 1:1 Trisomia 21Cortica<strong>le</strong>Demenza Amnestico, Parkinsonismo - 1:1 ApoE4Pugilistica Cortica<strong>le</strong>Demenza Amnestico, No - - BRI2familiare Cortica<strong>le</strong>In associazione con altre patologieDistrofia Fronta<strong>le</strong> No - 2:1 DMPKMiotonicaDegenerazione Ritardo menta<strong>le</strong> Disturbo del cammino, - PANK2PKANSegni extrapiramidaliNeumann-Pick Ritardo menta<strong>le</strong>, Distonia, Atassia, - NPC1,Psicosi Oftalmop<strong>le</strong>gia NPC2
I principali quadri clinici 11. Demenza fronto-tempora<strong>le</strong> con parkinsonismoassociate al cromosoma 17 (FTDP-17)Con il termine di “demenza fronto-tempora<strong>le</strong>” s’identifica un gruppo di sindromi cliniche(preva<strong>le</strong>ntemente sporadiche con esordio insidioso nella 6^ decade e decorsoprogressivo) caratterizzate dalla degenerazione circoscritta dei lobi pre-frontali e temporalianteriori con quadro neuropatologico non-Alzheimer.Tali forme sono attualmente suddivise in tre varianti:1. variante fronto-tempora<strong>le</strong> o comportamenta<strong>le</strong>,2. afasia progressiva non fluente,3 demenza semantica [7].La presenza di forme familiari <strong>le</strong>gate al cromosoma 17 (con trasmissione autosomicodominante) e sostenute da mutazioni della tau è stata originariamente descritta nel 1998[8], con successiva identificazione di circa 40 mutazioni in oltre cento famiglie.Il fenotipo <strong>clinico</strong> dei casi famigliari di FTDP-17 è generalmente simi<strong>le</strong> a quello dei casisporadici, con la presenza di segni parkinsoniani addizionali.2. Paralisi Sopranuc<strong>le</strong>are Progressiva (PSP)Originariamente descritta nel 1964 [9] è una condizione sporadica (rari casi familiari)con esordio insidioso dopo i 40 anni, decorso progressivo e preva<strong>le</strong>nza stimata intornoa 6.5/100.000 [10].In seguito è stata identificata come “taupatia” per la presenza d’inclusioni intracellularidi tau iperfosforilata a livello ippocampa<strong>le</strong>, mesencefalico (pallido, subtalamo,sostanza nera) e pontino con presenza di abbondanti degenerazioni neurofibrillari.Si manifesta con una sindrome parkinsoniana (preva<strong>le</strong>ntemente assia<strong>le</strong> e non-responsivaalla <strong>le</strong>vodopa), precoce instabilità postura<strong>le</strong> con cadute, oftalmop<strong>le</strong>gia vertica<strong>le</strong>,sintomi bulbari e disfunzione cognitiva.Tuttavia Williams e coll. [11] hanno documentato accanto alla presentazione più frequentee tipica (“sindrome di Richardson”) un fenotipo <strong>clinico</strong> con manifestazioni inizialisovrapponibili alla m. idiopatica di Parkinson.Le due forme differiscono per la composizione della tau e per l’associazione con il polimorfismoH1/H1.3. Degenerazione Cortico-Basa<strong>le</strong> (CBD)Descritta nel 1968 [12] è una condizione sporadica (rarissimi casi familiari) con esordioinsidioso a partire dalla 6 a decade e decorso progressivo.La preva<strong>le</strong>nza è incerta, ma probabilmente sottostimata per la variabilità fenotipicadella presentazione.E’ possibi<strong>le</strong>, infatti, una presentazione preva<strong>le</strong>ntemente motoria (sindrome rigido-acineticaasimmetrica, distonia, aprassia ideomotoria e segmenta<strong>le</strong> con “arto alieno”) o, piùraramente, cognitiva (ridotta fluenza verba<strong>le</strong>, alterazioni del comportamento, demenza).La variabilità fenotipica condiziona anche l’utilizzo di specifici criteri diagnostici [13].Il quadro neuropatologico presenta similarità con la PSP, ma si distingue per l’asimmetricitàdell’atrofia cortica<strong>le</strong> e la maggior presenza di placche astrocitiche e fibre argirofi<strong>le</strong>.Anche il profilo mo<strong>le</strong>colare della tau è analogo a quello riscontrato nella PSP [14].La sovrapposizione <strong>clinico</strong>-patologica e genetica suggerisce che PSP e CBD possanorappresentare fenotipi diversi di una patologia con background genetico comune [15].4. Le altre formeInclusioni tau-positive caratterizzano sul piano neuropatologico altre rare condizioni confenotipo variabi<strong>le</strong> motorio-cognitivo, quali: comp<strong>le</strong>sso parkinson-demenza di Guam,parkinsonismo post-enecefalitico, demenza pugilistica, neurodegenerazione associataalla pantotenato-chinasi [5].