Chem 459/559—Medicinal Chemistry Problem Set 3
Chem 459/559—Medicinal Chemistry Problem Set 3
Chem 459/559—Medicinal Chemistry Problem Set 3

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
<strong>Chem</strong> <strong>459</strong>/<strong>559—Medicinal</strong> <strong>Chem</strong>istry<br />
<strong>Problem</strong> <strong>Set</strong> 3<br />
Fall 2008<br />
Name________________________ 20 points<br />
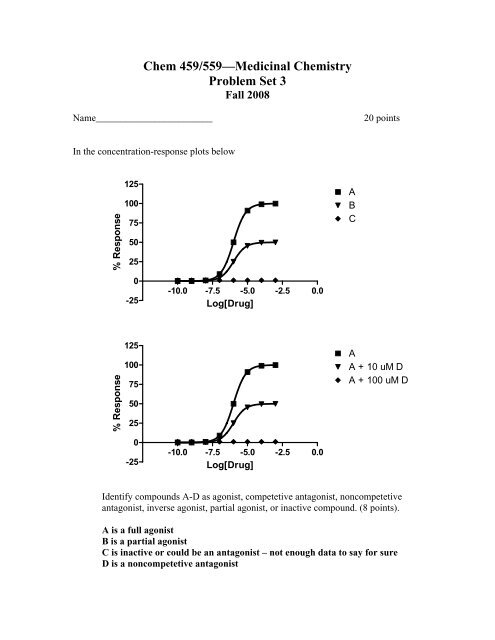
In the concentration-response plots below<br />
% Response<br />
% Response<br />
125<br />
100<br />
75<br />
50<br />
25<br />
0<br />
-25<br />
125<br />
100<br />
75<br />
50<br />
25<br />
0<br />
-25<br />
-10.0 -7.5 -5.0 -2.5 0.0<br />
Log[Drug]<br />
-10.0 -7.5 -5.0 -2.5 0.0<br />
Log[Drug]<br />
A<br />
B<br />
C<br />
A<br />
A + 10 uM D<br />
A + 100 uM D<br />
Identify compounds A-D as agonist, competetive antagonist, noncompetetive<br />
antagonist, inverse agonist, partial agonist, or inactive compound. (8 points).<br />
A is a full agonist<br />
B is a partial agonist<br />
C is inactive or could be an antagonist – not enough data to say for sure<br />
D is a noncompetetive antagonist
Draw acetylcholine and indicate all possible modes of interaction with its receptors (5<br />
points).<br />
H-bond<br />
acceptor<br />
π-bond<br />
O van-Der<br />
Waals<br />
CH3 can do<br />
charge transfer<br />
H3C O<br />
N<br />
+<br />
H-bond acceptor<br />
CH 3<br />
CH 3<br />
ion-ion or ion-dipole<br />
All indicated dipoles can participate in<br />
dipole-dipole and ion-dipole interactions<br />
(the bonds participate, not the individual atoms)<br />
Given that the carbonyl oxygen is required for interaction with nicotinic receptors and the<br />
ester oxygen is required for interaction with muscarinic receptors, suggest a potentially<br />
selective bioisosteric analog for nicotinic receptors and an analogous bioisostere that<br />
would be muscarinic selective (4 points).<br />
may be cyclic<br />
X CH3 X = S, CH2 O<br />
Muscarinic<br />
N<br />
CH 3<br />
CH 3<br />
may be cyclic<br />
X = O, N<br />
X CH 3<br />
Nicotinic<br />
Although acetylcholine has no chiral centers, draw a structure indicating how it might<br />
interact in a stereoselective fashion with a receptor based on conformations (3 points).<br />
O H CH3 O<br />
H<br />
H 3C O<br />
N<br />
CH 3<br />
CH 3<br />
H 3C O<br />
These are two examples. Any conformation in which the<br />
acetate or ammonium are not in the same plane (eclipsed syn or<br />
anti) are chiral conformations. All gauche conformers are<br />
chiral if conformation is restricted by binding to the receptor.<br />
H 3C<br />
N<br />
CH 3<br />
H<br />
H<br />
N<br />
CH 3<br />
CH 3<br />
CH 3
Modeling/conformational analysis exercise (20 points extra credit)<br />
To perform this exercise, you will need a model kit and will need to use the computers in<br />
room S51J, on which are installed <strong>Chem</strong>Draw and <strong>Chem</strong>3D.<br />
Using <strong>Chem</strong>Draw, sketch nicotine as shown at right (without numbering).<br />
Open <strong>Chem</strong>3D. Copy the structure from <strong>Chem</strong>Draw and paste it<br />
into <strong>Chem</strong>3D. Check to be sure the methyl group is oriented<br />
correctly according to the structure above. Rotate the molecule<br />
using the sliders on the right hand and bottom edges of the screen<br />
until you are confident you have the correct structure (Do not<br />
select any atoms or bonds any bonds yet.).<br />
Using a model kit, build a handheld model of nicotine. Attach hydrogens to all atoms<br />
using the 20 mm bonds (shorter grey).<br />
Move the cursor over an atom and leave it for a moment, a pop-up box will give a<br />
description of the atom and measurements associated with it. Likewise having the cursor<br />
over a bond will show the bond characteristics.<br />
Note the initial dihedral angle between the the aromatic ring and the CN bond of the<br />
pyrrolidine (you can do this by holding down the shift key and highlighting the four<br />
atoms indicated in the structure above – be careful not to select the lone pair on the<br />
nitrogen). From the Object menu select <strong>Set</strong> Dihedral Angle. A message box will open at<br />
the side showing the dihedral angle for the selected atoms (Do not enter a value – it will<br />
distort the structure). The initial angle should be near 0 o . If it is not, click and highlight<br />
the bond between the two rings (2 and 3 as shown above). Rotate the bond with the<br />
slider on the left until the dihedral angle in the window is nearly 0 o . Rotate the bond on<br />
your handheld model to match.<br />
From the MM2 menu, select Compute Properties and select Steric Energy Summary in the<br />
dialog box and click OK. A message will appear at the bottom of the screen with the<br />
Steric Energy. Write this number down here. 23.709 kcal/mol<br />
From the MM2 menu, select Minimize Energy and set the RMS gradient to 0.05 and click on<br />
Run to execute the minimization. This will relax the molecule into a low energy state for<br />
the pyrrolidine ring and the torsional energy about the bond connecting the two rings.<br />
Once you have done so, readjust the dihedral angle so that the angle is again very near 0 o .<br />
Write this number down in the left hand column of the table on the next page.<br />
(Note: initial energy 7.7-8.1 kcal/mol is at a dihedral angle of -28 o )<br />
Select and rotate the bond connecting the two rings by 30 o increments and calculate the<br />
associated energies using Compute Properties and plot them in the graph on the right.<br />
Each time, move your handheld model to the same position and observe the two in<br />
comparison.<br />
N<br />
1<br />
2<br />
3<br />
N 4<br />
CH 3
φ( o ) dieq nomin diax Cis<br />
0 8.5 23.7 9.2 210<br />
30 16.9 46.3 9.6 24.6<br />
60 25.7 51.9 9.1 9.7<br />
90 19.0 36.0 9.5 9.8<br />
120 8.5 21.9 15.4 25.7<br />
150 8.1 21.2 12.9 72.0<br />
180 9.6 23.6 10.2 179<br />
210 23.2 45.3 11.4 22.7<br />
240 23.5 52.1 9.8 9.4<br />
270 14.4 37.1 10.8 9.4<br />
300 8.4 22.2 23.8 24.9<br />
330 7.8 21.1 16.8 89.8<br />
M1 7.5 7.5 8.2 8.60<br />
M2 7.5 7.5 8.3 8.56<br />
Dieq - Values for trans diequatorial Nomin - Dieq Values w/o minimization<br />
Diax - Values for trans diaxial Cis - Values for cis<br />
Rotate the bond between the rings such that the dihedral angle is around 60 o . and<br />
minimize the structure. In the table, write down the dihedral angle (M1) and steric<br />
energy from the minimized structure.<br />
From the minimized structure, rotate the bond between the rings to -60 o and repeat the<br />
minimization. Record the dihedral angle (M2) and energy in the table.<br />
Based on your experiment, answer the following questions.<br />
Steric energy kcal/mol<br />
Compare your 0 o energy in the table with the value you wrote on the preceeding page. Is<br />
there any difference and if so, what would account for it (5 points)?<br />
The energy before minimization will be higher because the bond angles are<br />
arbitrary and have not been optimized.<br />
60<br />
50<br />
40<br />
30<br />
20<br />
10<br />
0<br />
0<br />
0 30 60 90 120 150 180 210 240 270 300 330 360<br />
Dihedral angle (degrees)<br />
Trans-nomin<br />
Trans-dieq<br />
Trans-diax<br />
Cis<br />
How well do the minimized structures agree with the graph you constructed? Are the<br />
dihedral angles and energies close to what you expected (5 points)?<br />
The minimized structure will have angles of about -55 o or +129 o for the backward<br />
and forward rotations respectively for the correct (trans-diequatorial) structure.<br />
This correlates nicely with the graph above, but the energies are better due to a full<br />
optimization of structure. The angles for the diaxial are (-97 o , +64 o ) and the cis (-<br />
103 o , +66 o ) will be different of course.<br />
From examination of your handheld model and/or the computer generated model, can<br />
you suggest a structural reason why the minimum energy conformations found should be<br />
preferred over the higher energy conformations seen (Be specific, 5 points)?<br />
In those conformations there is less interaction between the two rings due to<br />
coplanarity of the N-methyl and/or the C3 methylene of the pyrrolidine ring.<br />
250<br />
200<br />
150<br />
100<br />
50<br />
Steric energy (Cis only)