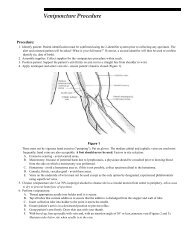
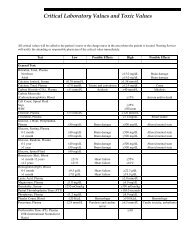
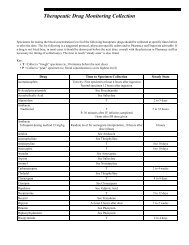
AIP rarely presents prior to puberty, with onset most commonly between ages 20 and 40. It is characterized by episodes of acute neuropathic symptoms. Most patients, approximately 95%, experience severe abdominal pain, often in conjunction with nausea, vomiting, and constipation. Peripheral neuropathy is common. However, given the extensive list of differential diagnoses for patients experiencing peripheral neuropathy, testing for AIP in the absence of abdominal pain rarely identifies AIP patients and is not recommended. Patients frequently display psychiatric symptoms presenting in the form of psychotic episodes, depression, and anxiety. Other features of an acute attack include circulatory disturbances such as hypertension and tachycardia. Dysuria and urinary retention, sometimes requiring catheterization, may be seen. Less frequently, patients may experience seizures, respiratory paralysis, fever, and diarrhea. Given the highly variable and nonspecific nature of the neurovisceral symptoms observed in AIP attacks, the condition is often overlooked in the clinical setting. Unfortunately, appropriate laboratory investigations are often not conducted, and many patients are misdiagnosed or become incorrectly labeled as narcotic seeking. This leads to a potentially life-threatening situation as patients continue to be at risk for an acute attack. Inheritance of AIP occurs in an autosomal dominant manner with reduced penetrance. Approximately 10-20% of individuals with a PBGD enzyme deficiency will become symptomatic during their lifetime, although some recent studies have questioned this low penetrance rate. 1,2 While the vast majority of patients will never exhibit symptoms, the identification of asymptomatic, affected individuals in families with known AIP is crucial. <strong>The</strong> diagnosis of asymptomatic patients allows for the avoidance of precipitating factors, thereby minimizing the risk of a life-threatening porphyric attack. With respect to the initial diagnosis of symptomatic patients believed to be in an acute AIP crisis, urine porphyrins (#8562 Porphyrins, Quantitative, Urine), ALA (#8406 Aminolevulinic Acid [ALA], Urine), and PBG (#82068 Porphobilinogen, Quantitative, Random, Urine) should be analyzed. Substantial financial savings and improvement in the appropriateness of testing can be attained by following our suggested testing strategies for the acute porphyrias (see Table 1). 5 This will ensure that another acute porphyria, with features similar to AIP, is not missed. PBGD (#9625 Aminolevulinic Acid Dehydratase [ALA-D] and Porphobilinogen Deaminase [Pbg-D] (Uroporphyrinogen Synthase [UpgS]), Erythrocytes) enzyme activity should be evaluated either in conjunction with these urine analyses or preferably in a stepwise fashion when indicated, based upon the urine studies. Identification of asymptomatic, affected family members, including children, is possible and requires either biochemical or molecular analysis. However, molecular testing is not readily available on a clinical basis at this time. Urinary ALA and PBG values are method dependent and can vary by institution. Some experts believe that these analytes will never fall within the normal range in asymptomatic, affected individuals, whereas others argue that these values can normalize in such patients. With the assays available through Mayo Medical Laboratories (MML), elevated urinary ALA and PBG values have been observed in asymptomatic individuals in whom AIP status was previously unknown. Provision of clinical information and reason for referral is important for accurate result interpretation. Regarding the diagnosis of asymptomatic infants and children, there is evidence that PBGD activity fluctuates considerably during the first 9-12 months of life; therefore, enzyme analysis should be performed after 1 year of age. In some cases, it is helpful to perform PBGD analysis of known affected family members when attempting to rule in/out AIP in asymptomatic relatives. Given that up to 10% of asymptomatic individuals with AIP will have a normal PBGD result, the urine assays are important diagnostic tools. Variegate Porphyria Variegate porphyria (VP) is an autosomal dominant acute porphyria that results from a reduction in the activity of protoporphyrinogen oxidase activity. VP is pan-ethnic, although high prevalence is reported in South Africa (3/1000 individuals) and Finland. Reduced penetrance is observed and symptoms very rarely present before puberty. Clinical presentation of VP is similar to other acute porphyrias, with symptoms including abdominal pain, vomiting, neuropathies, and psychiatric sequelae. However, cutaneous involvement is usually more pronounced. In fact, dermatologic manifestations in VP are very similar to those seen in PCT and include blistering, hyperpigmentation, and hypertrichosis of sun-exposed areas. Moreover, while neuropathic symptoms appear only during acute crisis, photosensitivity remains a chronic symptom. In rare instances, homozygous VP, with marked deficiency of protoporphyrinogen oxidase enzyme 6 11/02
activity, has been described. Patients typically present in early childhood with photosensitivity resulting in severe cutaneous manifestations, neurologic symptoms including seizures, and developmental delay. <strong>The</strong> diagnosis of VP relies upon porphyrin analysis in urine (#8562 Porphyrins, Quantitative, Urine) and feces (#81652 Porphyrins, Feces), as enzyme and molecular analysis of protoporphyrinogen oxidase are not readily available on a clinical basis. Depending upon whether the patient is experiencing an acute crisis or is asymptomatic, urine coproporphyrin, ALA, and PBG values are elevated to varying degrees. Values may be as high as 10-20 times normal during acute crises but may be normal or only mildly elevated between attacks. During crises, fecal porphyrin analysis shows coproporphyrin levels are, at a minimum, double with a coproporphyrin III to coproporphyrin I ratio in the 3-10 range (normal ratio