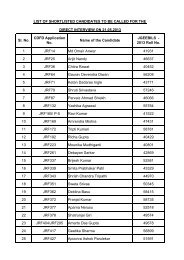
EMBnet course Proteomics using Bioinformatics tools
EMBnet course Proteomics using Bioinformatics tools
EMBnet course Proteomics using Bioinformatics tools

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
<strong>EMBnet</strong> <strong>course</strong><br />
<strong>Proteomics</strong> <strong>using</strong><br />
<strong>Bioinformatics</strong> <strong>tools</strong><br />
MS identification <strong>tools</strong><br />
Patricia M. Palagi<br />
PIG, SIB, Geneva<br />
PMP
The data: list of m/z values<br />
Peptide mass values and intensities<br />
MS<br />
840.6950 13.75<br />
1676.9606 26.1<br />
1498.8283 128.9<br />
1045.564 845.2<br />
2171.9670 2.56<br />
861.1073 371.2<br />
842.51458 53.7<br />
1456.7274 12.9<br />
863.268365 3.1<br />
Parent mass value<br />
fragment mass values<br />
MS/MS<br />
Parent mass charge<br />
1163.7008 2<br />
86.1105 220.1429<br />
86.1738 13.7619<br />
102.0752 4.3810<br />
147.1329 57.3333<br />
185.1851 649.0953<br />
185.3589 5.3810<br />
186.1876 81.4286<br />
213.0791 1.4286<br />
fragment intensities<br />
PMP
PMP<br />
The <strong>tools</strong>
One direct access to all- ExPASy<br />
http://www.expasy.org/<strong>tools</strong>/<br />
PMP
Automatic protein identification<br />
- Peptide mass fingerprinting – PMF<br />
- MS/MS sequence search<br />
- MS/MS spectra library search<br />
- MS/MS prospective analysis (tag,<br />
open mod, de novo sequencing<br />
PMP
Peptide mass fingerprinting = PMF<br />
MS database matching<br />
Protein(s)<br />
Enzymatic<br />
digestion<br />
…MAIILAGGHSVRFGPKAF<br />
AEVNGETFYSRVITLESTNM<br />
FNEIIISTNAQLATQFKYPN<br />
VVIDDENHNDKGPLAGIYTI<br />
MKQHPEEELFFVVSVDTPM<br />
ITGKAVSTLYQFLV …<br />
Sequence<br />
database entry<br />
Peptides<br />
In-silico<br />
digestion<br />
Mass spectra<br />
- MAIILAGGHSVR<br />
-FGPK<br />
- AFAEVNGETFYSR<br />
- VITLESTNMFNEIIISTNAQLATQFK<br />
- YPNVVIDDENHNDK<br />
…<br />
Theoretical<br />
proteolytic peptides<br />
Peaklist<br />
840.695086<br />
1676.96063<br />
1498.8283<br />
1045.564<br />
2171.967066<br />
861.107346<br />
842.51458<br />
1456.727405<br />
863.268365<br />
Match<br />
861.107346<br />
838.695086<br />
1676.96063<br />
1498.8283<br />
1045.564<br />
2171.967066<br />
842.51458<br />
1457.827405<br />
863.268453<br />
Theoretical<br />
peaklist<br />
Result:<br />
ranked list<br />
of protein<br />
candidates<br />
PMP
Peptide mass fingerprinting<br />
What you have:<br />
- Set of peptide mass values<br />
- Information about the protein: molecular weight, pI, species.<br />
- Information about the experimental conditions: mass spectrometer<br />
precision, calibration used, possibility of missed-cleavages, possible<br />
modifications<br />
- Biological characteristics: post-translational modifications, fragments<br />
What will do the tool:<br />
- Match between this information and a protein sequence database<br />
What will you get:<br />
- a list of probable identified proteins<br />
PMP
What is the expected information in a<br />
submission form?<br />
• Place to upload a spectrum (many spectra)<br />
• Description of the sample process used<br />
– Chemical process such as alkylation/reduction,<br />
– Cleavage properties (enzyme),<br />
– Mass tolerance (m/z tolerance)<br />
• Search space<br />
– Sequence databank,<br />
– taxonomy restriction<br />
– Mw, pI restriction<br />
• Scoring criteria and filters<br />
PMP
One example of parameter<br />
effects on the search<br />
• Accepted mass tolerance<br />
‣ due to imprecise measures and calibration problems<br />
PMP Source: Introduction to proteomics: <strong>tools</strong> for the new biology. Daniel C. Liebler. Human Press. 2002
Summary of PMF <strong>tools</strong><br />
Tool<br />
Aldente<br />
Mascot<br />
MS-Fit<br />
ProFound<br />
PepMAPPER<br />
PeptideSearch<br />
PepFrag<br />
Source website<br />
www.expasy.org/cgi-bin/aldente<br />
www.matrixscience.com/<br />
prospector.ucsf.edu/<br />
prowl.rockefeller.edu/profound_bin/WebProFound.exe<br />
wolf.bms.umist.ac.uk/mapper/<br />
www.mann.emblheidelberg.de/GroupPages/PageLink/peptidesearchpage.html<br />
prowl.rockefeller.edu/prowl/pepfragch.html<br />
PMP<br />
Non exhaustive list!
Scoring systems<br />
• Essential for the identification! Gives a confidence value to<br />
each matched protein<br />
• Three types of scores<br />
• Shared peaks count (SPC): simply counts the number of<br />
matched mass values (peaks)<br />
• Probabilistic scores: confidence value depends on<br />
probabilistic models or statistic knowledge used during the match<br />
(obtained from the databases)<br />
• Statistic-learning: knowledge extraction from the influence of<br />
different properties used to match the proteins (obtained from the<br />
databases)<br />
PMP
Mascot<br />
http://www.matrixscience.com/<br />
• Internet free version in the above website<br />
(commercial versions available too)<br />
•Choice of several databases.<br />
• Considers multiple chemical modifications.<br />
• 0 to 9 missed-cleavages.<br />
• Score based on a combination of probabilistic<br />
and statistic approaches (is based on Mowse<br />
score).<br />
• Considers Swiss-Prot annotations for Splice<br />
Variants (in locally installed versions).<br />
PMP
Mascot - principles<br />
• Probability-based scoring<br />
• Computes the probability P that a match is<br />
random<br />
• Significance threshold p< 0.05 (accepting that<br />
the probability of the observed event occurring<br />
by chance is less than 5%)<br />
• The significance of that result depends on the<br />
size of the database being searched.<br />
• Mascot shades in green the insignificant hits<br />
• Score: -10Log 10 (P)<br />
PMP
Mascot<br />
Input<br />
PMP
Decoy<br />
Output<br />
Hints about the significance<br />
of the score<br />
PMP
Sequence coverage<br />
Output<br />
Peptides matched<br />
Error<br />
function<br />
PMP
Aldente<br />
• SwissProt/TrEMBL db, indexed masses (trypsine and many<br />
others).<br />
• Considers chemical modifications and user specified<br />
modifications.<br />
• Considers biological modifications (annotations SWISS-PROT).<br />
• 0 or 1 missed-cleavages.<br />
• Use of robust alignment method (Hough transform):<br />
• Determines deviation function of spectrometer<br />
• Resolves ambiguities<br />
• Less sensitive to noise<br />
PMP
Aldente – summary<br />
Experimental masses / peaks<br />
Spectrometer<br />
calibration error<br />
Spectrometer<br />
internal error<br />
• The Hough Transform estimates from<br />
the experimental data the deviation<br />
function of the mass spectrometer (the<br />
calibration error function).<br />
Theoretical masses / peptides<br />
• The program optimizes the set of<br />
best matches, excluding noise and<br />
outliers, to find the best alignment.<br />
PMP
PMP<br />
Aldente - Input
PMP<br />
Aldente - Input
PMP<br />
Aldente - Input
PMP<br />
Aldente - Input
PMP<br />
Aldente - Input
Output<br />
Hints about<br />
the<br />
significance of<br />
the score<br />
PMP
PMP<br />
Information from Swiss-Prot<br />
annotation. Processed protein (signal<br />
peptide is cleaved).
PMP<br />
BioGraph
What is the expected information<br />
in an identification result?<br />
• A summary of the search parameters<br />
• A list of potentially identified proteins (AC numbers) with<br />
scores and other evidences<br />
• A detailed list of potentially identified peptides (associated or<br />
not to the potentially identified proteins) with scores<br />
• Possibilities to validate/invalidate the provided results (info on<br />
the data processing, on the statistics, links to external<br />
resources, etc.)<br />
• Possibilities to export the (validated) data in various formats<br />
PMP
Protein characterization with PMF<br />
data<br />
1 protein entry<br />
does not represent<br />
1 unique molecule<br />
- Exact primary structure<br />
- Splicing variants<br />
- Sequence conflicts<br />
-PTMs<br />
Characterization <strong>tools</strong> at ExPASy <strong>using</strong> peptide mass fingerprinting data<br />
http://www.expasy.org/<strong>tools</strong>/<br />
FindMod<br />
GlycoMod<br />
FindPept<br />
Prediction <strong>tools</strong><br />
• PTMs and AA substitutions<br />
• Oligosaccharide structures<br />
• Unspecific cleavages<br />
PMP
PMP<br />
SWISS-PROT feature table:<br />
active protein is more than just translation of<br />
gene sequence (example: P20366)
PMP<br />
Detection of PTMs in MS<br />
769.8<br />
893.4<br />
1326.7<br />
1501.9<br />
2100.6<br />
1056.1<br />
624.3<br />
624.3<br />
769.8<br />
893.4<br />
994.5<br />
994.5<br />
1056.1<br />
1326.7<br />
1501.9<br />
1759.8<br />
1759.8 1923.4<br />
1923.4<br />
2100.6<br />
Unmodified<br />
tryptic<br />
masses<br />
600 2200<br />
Δ m/z<br />
=> PTM<br />
769.8<br />
769.8<br />
893.4<br />
893.4<br />
1056.1<br />
1070.1<br />
1326.7<br />
1326.7<br />
1501.9<br />
1501.9<br />
2100.6<br />
2100.6<br />
624.3<br />
624.3<br />
994.5<br />
994.5<br />
1759.8<br />
1759.8 1923.4 1923.4<br />
Tryptic<br />
masses of<br />
a modified<br />
protein<br />
600 2200
PMP<br />
FindMod http://www.expasy.org/<strong>tools</strong>/findmod/<br />
AA modifications<br />
DB entry<br />
experimental<br />
options<br />
experimental<br />
masses
FindMod Output<br />
}unmodified peptides,<br />
modified peptides<br />
known in SWISS-PROT<br />
and chemically modified<br />
peptides<br />
}<br />
putatively modified<br />
peptides predicted<br />
by mass differences<br />
+ putative AA substitutions<br />
PMP
Modification rules can be defined from<br />
SWISS-PROT, PROSITE and the literature<br />
some examples:<br />
modification amino acid rule exceptions<br />
farnesylation Cys -<br />
palmitoylation Cys Ser, Thr<br />
O-GlcNAc Ser, Thr Asn<br />
amidation<br />
pyrrolidone carboxylic acid Gln (N-term) -<br />
Xaa (C-term) where Gly followed Xaa<br />
phosphorylation in eukaryotes: Ser, Thr, Asp, His, Tyr -<br />
in prokaryotes: Ser, Thr, Asp, His, Cys -<br />
PMP<br />
sulfatation in eukaryotes<br />
Tyr, PROSITE PDOC00003
FindMod Output - Application of Rules<br />
- potentially modified peptides that agree with rules are listed<br />
- amino acids that potentially carry modifications are shown<br />
- peptides potentially modified only by mass difference<br />
PMP<br />
- predictions can be tested by MS-MS peptide fragmentation
FindPept<br />
http://www.expasy.org/<strong>tools</strong>/findpept.html<br />
• From MS (peptide mass fingerprint) data -<br />
detection of :<br />
– Matching peptides for unspecific cleavage<br />
– Masses resulting from possible contaminants<br />
– Matching peptides for specific cleavage (16 different<br />
enzymes)<br />
– Peptides resulting from protease autolysis<br />
PMP
PMP<br />
FindPep
PMP<br />
FindPep
PMP<br />
FindPep
PMP<br />
FindPep
MS/MS based identification <strong>tools</strong><br />
• Tag search- Tools that search peptides based on a MS/MS<br />
Sequence Tag<br />
– MS-Tag and MS-Seq, PeptideSearch<br />
• Ion search or PFF - Tools that match MS/MS experimental<br />
spectra with “theoretical spectra” obtained via in-silico<br />
fragmentation of peptides generated from a sequence<br />
database<br />
– Phenyx, Mascot, Sequest, X!Tandem, OMSSA, ProID, …<br />
• de novo sequencing - Tools that directly interpret MS/MS<br />
spectra and try to deduce a sequence<br />
– Convolution/alignment (PEDENTA)<br />
– De-novo sequencing followed by sequence matching<br />
(Peaks, Lutefisk, Sherenga, PeptideSearch)<br />
– Guided Sequencing (Popitam)<br />
In all cases, the output is a peptide structure per MS/MS spectrum<br />
PMP
Peptide fragmentation<br />
fingerprinting = PFF = ion search<br />
MS/MS database matching<br />
Protein(s)<br />
…MAIILAGGHSVRFGPKAF<br />
AEVNGETFYSRVITLESTNM<br />
FNEIIISTNAQLATQFKYPN<br />
VVIDDENHNDKGPLAGIYTI<br />
MKQHPEEELFFVVSVDTPM<br />
ITGKAVSTLYQFLV …<br />
PMP<br />
Enzymatic<br />
digestion<br />
Sequence<br />
database entry<br />
In-silico<br />
digestion<br />
Peptides<br />
- MAIILAGGHSVR<br />
-FGPK<br />
- AFAEVNGETFYSR<br />
- VITLESTNMFNEIIIK<br />
- YPNVVIDDENNDK<br />
…<br />
Theoretical<br />
proteolytic peptides<br />
MS/MS spectra<br />
of peptides<br />
In-silico<br />
fragmentation<br />
-MAIILAG<br />
-MAIILA<br />
-MAIIL<br />
-MAII<br />
-MAI<br />
-M<br />
-M<br />
-AIILAG<br />
Theoretical<br />
fragmented<br />
peptides<br />
Ions peaklists<br />
340.695086<br />
676.96063<br />
498.8283<br />
545.564<br />
1171.967066<br />
261.107346<br />
342.51458<br />
456.727405<br />
363.268365<br />
Match<br />
361.107346<br />
338.695086<br />
676.96063<br />
498.8283<br />
1045.564<br />
1171.967066<br />
342.51458<br />
457.827405<br />
263.268453<br />
Theoretical<br />
peaklist<br />
Result:<br />
ranked list<br />
of peptide<br />
and<br />
protein<br />
candidates
Ion-types<br />
offset<br />
-28<br />
-45<br />
-46<br />
0<br />
-17<br />
-18<br />
+17<br />
+28<br />
+ 2<br />
-15<br />
-16<br />
-15<br />
P' nterm<br />
P' cterm<br />
It is very important to know the ionic series<br />
produced by a spectrometer, otherwise<br />
potential matches will be missed.<br />
In the other hand, if an ion-type not present<br />
in the original spectrum is taken into account,<br />
it will contribute to get false positive<br />
matches.<br />
[N] is the mass of the N-term group<br />
[M] is the mass of the sum of the neutral amino acid residue<br />
masses<br />
PMP
Some PFF <strong>tools</strong><br />
Same principle of a PMF, but <strong>using</strong> MS/MS spectra<br />
Software<br />
InsPecT<br />
Mascot<br />
MS-Tag and MS-Seq<br />
PepFrag<br />
Phenyx<br />
Popitam<br />
ProID (download)<br />
Sequest*<br />
Sonar<br />
SpectrumMill*<br />
VEMS<br />
X!Tandem (download)<br />
Source website<br />
peptide.ucsd.edu/inspect.py<br />
www.matrixscience.com/search_form_select.html<br />
prospector.ucsf.edu<br />
prowl.rockefeller.edu/prowl/pepfragch.html<br />
phenyx.vital-it.ch<br />
www.expasy.org/<strong>tools</strong>/popitam<br />
sashimi.sourceforge.net/software_mi.html<br />
fields.scripps.edu/sequest/index.html<br />
65.219.84.5/service/prowl/sonar.html<br />
www.home.agilent.com<br />
www.bio.aau.dk/en/biotechnology/vems.htm<br />
www.thegpm.org/TANDEM<br />
PMP<br />
*Commercialized<br />
Non exhaustive list!
PMP
PMP<br />
Phenyx
Submission<br />
The Phenyx Web Interface:<br />
One result, multiple views<br />
Desktop<br />
Results<br />
views<br />
Results comparison<br />
Management console<br />
PMP P.A. Binz<br />
Excel, xml and text exports
The Proteins overview<br />
List of identified<br />
proteins<br />
Protein group<br />
description<br />
Corresponding list of<br />
identified peptides<br />
PMP P.A. Binz
The Proteins overview<br />
Hints about the<br />
significance of<br />
the score<br />
PMP
Hints about the<br />
significance of<br />
the score<br />
PMP<br />
Better when high intensity peaks are matched and ion series are<br />
extended, without too many and too big holes
The scoring system in Phenyx<br />
• The score is the sum of up to 12 basic scores such as:<br />
– presence of a, b, y, y++, B-H 2<br />
O…; co-occurrence of ion series<br />
(<strong>using</strong> HMMs), peak intensities, residue modifications (PTM or<br />
chemical), …<br />
• True probabilistic approach for each peptide match<br />
(likelihood of being correct)<br />
Search in a query database<br />
log --------------------------------<br />
(likelihood of being random)<br />
Search in a randomized set<br />
of peptides<br />
• Function of instruments and molecular types<br />
– Esquire 3000+, LCQ; iTRAQ vs. unmodified peptides<br />
• Scores are normalised into z-scores<br />
PMP
X!Tandem<br />
PMP<br />
www.thegpm.org
X!Tandem - output<br />
1<br />
2<br />
3<br />
PMP
The two-rounds search<br />
Mascot, Phenyx and X!Tandem<br />
The identification process may be launched in 2-rounds<br />
• Each round is defined with a set of search criteria<br />
– First round searches the selected database(s) with<br />
stringent parameters,<br />
– Second round searches the proteins that have<br />
passed the first round (relaxed parameters):<br />
⇒Accelerate the job when looking for many variable modifications,<br />
or unspecific cleavages<br />
⇒Appropriate when the first round defines stringent criteria to<br />
capture a protein ID, and the second round looks for looser<br />
peptide identifications<br />
PMP
Example 2 nd round<br />
1rnd,<br />
Only 3 fixed mods<br />
131 valid,<br />
75% cov.<br />
2rnd,<br />
Add variable mods<br />
205 valid,<br />
84% cov.<br />
2rnd,<br />
With all mods<br />
And half cleaved<br />
348 valid,<br />
90% cov.<br />
PMP
Source of errors in assigning<br />
peptides<br />
• Scores not adapted<br />
• Parameters are too stringent or too loose<br />
• Low MS/MS spectrum quality (many noise peaks, low<br />
signal to noise ratio, missing fragment ions, contaminants)<br />
• Homologous proteins<br />
• Incorrectly assigned charge state<br />
• Pre-selection of the 2 nd isotope (the parent mass is shifted<br />
of 1 Da. A solution is to take the parent mass tol. larger, but<br />
may drawn the good peptide too)…<br />
• Novel peptide or variant<br />
PMP
Hints to know when the<br />
PMP<br />
With MS<br />
identification is correct<br />
• Good sequence coverage: the larger the sub-sequences and the<br />
higher the sequence coverage value, the better<br />
• Consider the length of the protein versus the number of<br />
matched theoretical peptides<br />
•Better when high intensity peaks have been used in the identification<br />
•Scores: the higher, the better. The furthest from the 2nd hit the<br />
better<br />
• Filter on the correct species if you know it (reduces the search<br />
space, time, and errors)<br />
• Better when the errors are more or less constants among all<br />
peptides found.<br />
•If you have time, try many <strong>tools</strong> and compare the results
With MS/MS<br />
Hints to know when the<br />
identification is correct<br />
• The higher the number of peptides identified per protein, the better<br />
• Sequence coverage: the larger the sub-sequences and the higher<br />
the sequence coverage value, the better<br />
•Depends on the sample complexity and experiment workflow<br />
• Scores: the higher, the better.<br />
• Filter on the correct species if you know it (reduces the search<br />
space, time, and errors)<br />
• Better when high intensity peaks are matched and ion series are<br />
extended, without too many and too big holes.<br />
• Better when the errors are more or less constants among all ions.<br />
• If you have time, try many <strong>tools</strong> and compare the results<br />
PMP
E-values<br />
• For a given score S, it indicates the number of<br />
matches that are expected to occur by chance<br />
in a database with a score at least equal to S.<br />
• The e-value takes into account the size of the<br />
database that was searched. As a consequence<br />
it has a maximum of the number of sequences<br />
in the database.<br />
• The lower the e-value, the more significant the<br />
score is.<br />
PMP<br />
• An e-value depends on the calculation of the p-<br />
value.
p-value<br />
• A p-value describes the probability, which<br />
assesses the chance of validly rejecting the null<br />
hypothesis. If the p-value is 10 -5 then the<br />
rejection of the null hypothesis is due to chance<br />
with a probability of 10 -5 .<br />
• A p-value has a maximum of 1.0.<br />
• The larger the search space, the higher the p-<br />
value since the chance of a peptide being a<br />
random match increases.<br />
• The lower the p-Value, the more significant is the<br />
match.<br />
PMP Source: Lisacek, Practical <strong>Proteomics</strong>, 2006 Sep;6 Suppl 2:22-32
Z-score<br />
• Z-score is a dimensionless quantity<br />
derived by subtracting the population<br />
mean from an individual (raw) score and<br />
then dividing the difference by the<br />
population standard deviation.<br />
Z − score =<br />
• The z score reveals how many units of<br />
the standard deviation a case is above or<br />
below the mean.<br />
x<br />
− μ<br />
σ<br />
PMP Source: wikipedia
So what?<br />
• For small (significant) p-values, p and e are<br />
approximately equal, so the choice of one or the<br />
other is often equivalent. It is therefore<br />
reasonable to assimilate low p-values in Phenyx<br />
to e-values. X!Tandem simply switches e-values<br />
to log values to remove the powers of 10<br />
• For a single search (or set of sampled peptides),<br />
you can compare z-scores. However, when two or<br />
more searches are performed on different size<br />
spaces, you first need to look at the p-values<br />
before comparing z-scores.<br />
PMP Source: Lisacek, Practical <strong>Proteomics</strong>, 2006 Sep;6 Suppl 2:22-32