b) NMR: prediction of molecular alignment from structure
b) NMR: prediction of molecular alignment from structure
b) NMR: prediction of molecular alignment from structure

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
© 2008<br />
Nature<br />
Publishing<br />
Group<br />
http:<br />
/ / www.<br />
nature.<br />
com/<br />
natureprotocols<br />
PROTOCOL<br />
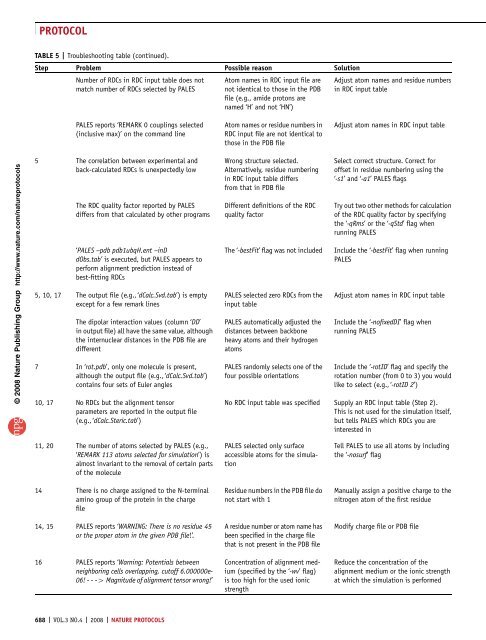
TABLE 5 | Troubleshooting table (continued).<br />
Step Problem Possible reason Solution<br />
Number <strong>of</strong> RDCs in RDC input table does not<br />
match number <strong>of</strong> RDCs selected by PALES<br />
PALES reports ‘REMARK 0 couplings selected<br />
(inclusive max)’ on the command line<br />
5 The correlation between experimental and<br />
back-calculated RDCs is unexpectedly low<br />
The RDC quality factor reported by PALES<br />
differs <strong>from</strong> that calculated by other programs<br />
‘PALES –pdb pdb1ubqH.ent –inD<br />
dObs.tab’ is executed, but PALES appears to<br />
perform <strong>alignment</strong> <strong>prediction</strong> instead <strong>of</strong><br />
best-fitting RDCs<br />
5, 10, 17 The output file (e.g., ‘dCalc.Svd.tab’) is empty<br />
except for a few remark lines<br />
The dipolar interaction values (column ‘DD’<br />
in output file) all have the same value, although<br />
the internuclear distances in the PDB file are<br />
different<br />
7 In ‘rot.pdb’, only one molecule is present,<br />
although the output file (e.g., ‘dCalc.Svd.tab’)<br />
contains four sets <strong>of</strong> Euler angles<br />
10, 17 No RDCs but the <strong>alignment</strong> tensor<br />
parameters are reported in the output file<br />
(e.g., ‘dCalc.Steric.tab’)<br />
11, 20 The number <strong>of</strong> atoms selected by PALES (e.g.,<br />
‘REMARK 113 atoms selected for simulation’) is<br />
almost invariant to the removal <strong>of</strong> certain parts<br />
<strong>of</strong> the molecule<br />
14 There is no charge assigned to the N-terminal<br />
amino group <strong>of</strong> the protein in the charge<br />
file<br />
14, 15 PALES reports ‘WARNING: There is no residue 45<br />
or the proper atom in the given PDB file!’.<br />
16 PALES reports ‘Warning: Potentials between<br />
neighboring cells overlapping, cut<strong>of</strong>f 6.000000e-<br />
06! ---4 Magnitude <strong>of</strong> <strong>alignment</strong> tensor wrong!’<br />
688 | VOL.3 NO.4 | 2008 | NATURE PROTOCOLS<br />
Atom names in RDC input file are<br />
not identical to those in the PDB<br />
file (e.g., amide protons are<br />
named ‘H’ and not ‘HN’)<br />
Atom names or residue numbers in<br />
RDC input file are not identical to<br />
those in the PDB file<br />
Wrong <strong>structure</strong> selected.<br />
Alternatively, residue numbering<br />
in RDC input table differs<br />
<strong>from</strong> that in PDB file<br />
Different definitions <strong>of</strong> the RDC<br />
quality factor<br />
Adjust atom names and residue numbers<br />
in RDC input table<br />
Adjust atom names in RDC input table<br />
Select correct <strong>structure</strong>. Correct for<br />
<strong>of</strong>fset in residue numbering using the<br />
‘-s1’ and‘-a1’ PALES flags<br />
Try out two other methods for calculation<br />
<strong>of</strong> the RDC quality factor by specifying<br />
the ‘-qRms’ orthe‘-qStd’ flag when<br />
running PALES<br />
The ‘-bestFit’ flag was not included Include the ‘-bestFit’ flag when running<br />
PALES<br />
PALES selected zero RDCs <strong>from</strong> the<br />
input table<br />
PALES automatically adjusted the<br />
distances between backbone<br />
heavy atoms and their hydrogen<br />
atoms<br />
PALES randomly selects one <strong>of</strong> the<br />
four possible orientations<br />
Adjust atom names in RDC input table<br />
Include the ‘-n<strong>of</strong>ixedDI’ flag when<br />
running PALES<br />
Include the ‘-rotID’ flag and specify the<br />
rotation number (<strong>from</strong> 0 to 3) you would<br />
like to select (e.g., ‘-rotID 2’)<br />
No RDC input table was specified Supply an RDC input table (Step 2).<br />
This is not used for the simulation itself,<br />
but tells PALES which RDCs you are<br />
interested in<br />
PALES selected only surface<br />
accessible atoms for the simulation<br />
Residue numbers in the PDB file do<br />
not start with 1<br />
A residue number or atom name has<br />
been specified in the charge file<br />
that is not present in the PDB file<br />
Concentration <strong>of</strong> <strong>alignment</strong> medium<br />
(specified by the ‘-wv’ flag)<br />
is too high for the used ionic<br />
strength<br />
Tell PALES to use all atoms by including<br />
the ‘-nosurf’ flag<br />
Manually assign a positive charge to the<br />
nitrogen atom <strong>of</strong> the first residue<br />
Modify charge file or PDB file<br />
Reduce the concentration <strong>of</strong> the<br />
<strong>alignment</strong> medium or the ionic strength<br />
at which the simulation is performed