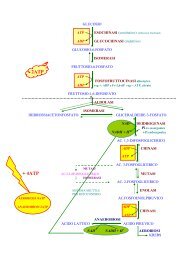
genetica3.pdf
genetica3.pdf
genetica3.pdf

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
Talassemia intermedia: sintomatico, ma non richiede trasfusione.<br />
Due mutazioni lievi<br />
Una mutazione molto lieve + α-talassemia o persistenza di HbF<br />
Talassemia maior: sintomatologia grave, richiede<br />
trasfusioni.<br />
Omozigote β 0 β 0<br />
Eterozigote β 0 β +<br />
La mutazione dell’emoglobina E<br />
30<br />
Mutazioni nel promotore del gene β-globinico che danno origine a talassemia-β + :<br />
Questa mutazione dà origine sia ad un’anomalia quantitativa, dovuta all’attivazione di un sito donatore di splicing criptico, sia<br />
ad una qualitativa, poiché il messaggero maturo corretto contiene una lisina al posto di un acido glutammico, al codone 26. La A al<br />
codone 26 è, almeno in apparenza, sufficiente ad attivare questo sito donatore di splicing alternativo, che viene quindi utilizzato il 40%<br />
delle volte.<br />
Meccanismi molecolari responsabili di HPFH<br />
• In A sono mostrate 3 delezioni HPFH che rimuovono<br />
grandi segmenti di DNA all’estremità 3’ del cluster βglobinico,<br />
comprendente i geni dell’adulto. Tuttavia,<br />
non tutte le delezioni di questo tipo sono responsabili di<br />
HPFH, come mostrato dalle 3 mutazioni δβ-talassemia<br />
rappresentate.<br />
• In B sono indicate le localizzazioni di mutazioni<br />
(numerate rispetto al sito di capping) nel promotore dei<br />
geni G γ e A γ delle globine fetali, riscontrate in individui<br />
con HPFH (indicati con la freccia sopra la linea).<br />
Queste posizioni presumibilmente indicano la posizione<br />
di importanti sequenze di controllo, ma nella maggior<br />
parte dei casi non coinvolgono i comuni elementi del<br />
promotore (CACCC, CCAAT o TATA). La barra al di<br />
sotto della sequenza indica la posizione di una piccola<br />
delezione in un paziente, che rimuove il secondo<br />
CCAAT box.<br />
MECCANISMO CONSEGUENZA