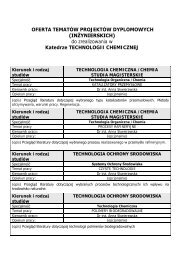
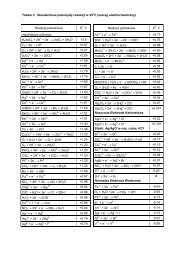
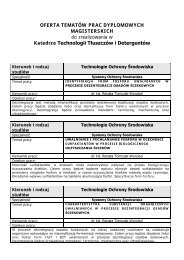
Slajdy z notatkami
Slajdy z notatkami
Slajdy z notatkami

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Przyczyny zjawiskpowierzchniowychW fazach skondensowanych pomiędzy cząsteczkami istnieją silne oddziaływaniaprzyciągające. W pobliŜu powierzchni (granicy faz) są one jednakniezrównowaŜone. Fakt ten jest przyczyną istnienia szeregu zjawisk na granicyfaz, m. in. napięcia powierzchniowego (międzyfazowego) i adsorpcji.oddziaływaniazrównowaŜoneCząsteczki w warstwie przypowierzchniowejznajdują się w odmiennym stanie energetycznymniŜ cząsteczki we wnętrzu fazy(energia powierzchniowa).Chem. Fiz. TCH II/23 11
Napięcie powierzchnioweJeŜeli cząsteczki we wnętrzu fazy skondensowanej sa ruchome (ciecz), tozrównowaŜenie sił we wnętrzu fazy i ich niezrównowaŜenie przy powierzchni sąuśrednione w czasie. Składowa siła przy powierzchni będzie zatem równieŜpowodować „wciąganie” cząsteczek przypowierzchniowych w głąb fazy ciekłej.Występować będzie zatem tendencja do samorzutnej minimalizacji powierzchnigranicy międzyfazowej.oddziaływanianiezrównowazone(silna składowa kuwnętrzu fazy)Chem. Fiz. TCH II/23 22
Napięcie powierzchniowe (2a)Przy danej objętości fazy ciekłej, bryłą o najmniejszej powierzchni jest kula.2Ak 4πr3Kula o promieniu r:= =4 3Vk3 π r rSześcian o tej samejobjętości, co kula, krawędź a:4 3= r ⇒ a =3 4π3π ⋅r2A a 63 16 2 2rs6 9π1 3, 724,53 16= = =4 3 4 3Vsr r9 π=3π3πr ra 2π ⋅rProstopadłościan o tej samej objętości,co kula, krawędzie a, 2a, 3a:Ap 22 π ⋅ r13=πVrpa2 3 4 2 222a811 4,16,53 4==4 3r r81=4 33π3πrChem. Fiz. TCH II/23 333= 3 9Wnioski: 1) im mniejsza objętość bryły (mniejszy wymiar liniowy), tym większystosunek A/V2) wartość ta granicznie wynosi3
Napięcie powierzchniowe (2b)Generalnie (dla kaŜdego typu bryły):2Ak 4πr3Kula o promieniu r:= =4 3Vk3 π r rSześcian o krawędzi a=2r(opisany na kuli):AV2 2s3s6a=3a24r=38rWnioski: 1) im mniejsza objętość bryły (mniejszy wymiar liniowy), tym większystosunek A/V (rośnie do nieskończoności); stosunek A/V jest w powyŜszymprzykładzie taki sam, ale V s >V k, (konkretnie V s ≈1,91V k ),2) stosunek A/V jest niezmiernie waŜny w procesach przebiegającychw układach wielofazowych (heterogenicznych). Albo zachodzą onena granicy faz, albo konieczny jest transport substratów/produktówprzez tę granicę.Chem. Fiz. TCH II/23 4=r4
Napięcie powierzchniowe (3)Dalsze wnioski:1) przy nieobecności sił zewnętrznych, fragmenty fazy ciekłej (krople)przyjmować będą samorzutnie kształt kulisty.2) ta sama objętość w postaci wielu bryłek (kropel), będzie miała większąpowierzchnię niŜ jedna duŜa kropla nA n >A 1 ,3) Ŝeby zwiększyć stosunek A/V (odkształcić kroplę) trzeba zadziałać siłązewnętrzną (z dokonaniem przesunięcia – zatem wykonać pracę)Zjawisko wskazane w punkcie 1 wykorzystywane jest do otrzymywania kulekdoskonałych ( w warunkach niewaŜkości i bez ruchu powietrza), a w warunkachziemskich w tzw. wieŜach śrutowych.Stosunek wielkości wskazanych w punkcie 2 (nA n /A 1 ) dla pojedynczej kroplio_średnicy 1 cm i n kropel o średnicy 1µm wynosi 10000 (tyle m 2 ile pojedynczakropla cm 2 ).Chem. Fiz. TCH II/23 5Pierwsze eksperymenty ad 1 robiono na stacji Skylab juŜ w maju 1973!5
Napięcie powierzchniowe (4)JeŜeli wyobrazimy sobie, Ŝe znad powierzchni cieczy wyciągamy poziomobeleczkę (fizycznie wymaga to konstrukcji pewnej ramki – na rysunku niepokazanej), to – o ile ciecz przylegać będzie do beleczki – wyciągniemy wraz znią film cieczy. Zakładając niewaŜkość beleczki, pomijając jej opory tarcia wramce oraz masę samego filmu (co zresztą moŜna uwzględnić w odpowiednichprocedurach kalibracyjnych), wykonana zostaniepraca przeciwko siłom dąŜącym do zmniejszeniapowierzchni granicy faz, poniewaŜ wyciągając filmją zwiększamy. Stosunek wykonanej pracy doprzyrostu tej powierzchni wynosi:wA=Fh2lh=F2l⎡ J= σ ⎢⎣m⎤⎥⎦⎡≡ ⎢⎣NmChem. Fiz. TCH II/23 62⎤⎥⎦W mianowniku jest 2, poniewaŜ film ma dwie strony.6
Napięcie powierzchniowe.Definicjeσ =wAFh=2lhF=2l⎡ J⎢⎣m⎤ ⎡ N ⎤⎥ ≡ ⎢ ⎥⎦ ⎣m⎦Wielkość σ nazywamy napięciem powierzchniowym i definiujemy jako pracęniezbędną do zwiększenia powierzchni granicy międzyfazowej o jednostkę.MoŜemy teŜ zauwaŜyć, Ŝe siły napięcia powierzchniowego zmniejszającepowierzchnię, F σ , działają stycznie do powierzchni filmu równowaŜąc siłę F.FF σ F σMoŜemy zatem zdefiniować napięcie powierzchniowe równieŜjako siłę styczną do powierzchni cieczy w kierunku prostopadłymdo liniowego przekroju powierzchni cieczy, przypadającąna jednostkę długości granicy międzyfazowej (tutaj równej 2l).W warunkach T,P = const, przy braku innych rodzajów pracywymienianej w układzie (np. objętościowej), moŜna zapisać:2dw ⎛ ∂G⎞σ = = ⎜ ⎟dA ⎝ ∂A⎠Chem. Fiz. TCH II/23 P,T 7Tu poprawnie narysowano styk filmu cieczy z beleczką. Poza tym dG jestujemne (kurczenie się powierzchni jest samorzutne), ale wtedy równieŜ dAma znak ujemny. Stąd nap. pow. ma znak dodatni.Dawne jednostki napięcia powierzchniowego (jeszcze są w tablicach) todyny/cm. 1 dyna/cm= 1 mN/m.7
Napięcie powierzchniowe.Metody pomiaru (1)Na podobnej do „ramki” zasadzie oparte są inne metody wyznaczania napięciapowierzchniowego. Są to róŜnego rodzaju wagi, przypominające nieco wagęArchimedesa. Najczęściej pomiar prowadzi się do momentu zerwania filmu,cały czas rejestrują siłę wyciągającą zanurzony w cieczy element pomiarowy.MoŜna zatem obliczyć róŜnicę sił tuŜ przed zerwaniem filmu i zaraz po, któraodpowiada wyłącznie siłom napięcia powierzchniowego działającym na obwodziepierwotnie częściowo zanurzonego elementu pomiarowego. Zwróćmyuwagę, Ŝe w tym sposobie (obecnie umoŜliwiającym bardzo precyzyjne pomiarydzięki skomputeryzowaniu układów pomiarowych) eliminuje się tarcie (prowadnicesą zbędne przy precyzyjnym podwieszeniu elementu pomiarowego),cięŜar elementu pomiarowego, a takŜe (zmienną) siłę jego wyporu.Elementy pomiarowe mają róŜne kształty, np. prostopadłościanu (blaszki),walca, lub torusa (w tym ostatnim przypadku mówimy o tzw. wadze Nouya).Chem. Fiz. TCH II/23 88
Napięcie powierzchniowe.Metody pomiaru (2)W przypadku prostopadłościanu (blaszki, 1):W przypadku walca (2):∆Fσ =πdσ =∆F2(a + b)W przypadku torusa (wagi Nouya, 3):ad∆Fσ =2πDb1) 2) 3)DChem. Fiz. TCH II/23 99
Napięcie międzyfazowe.Kąt zwilŜaniaNapięcie powierzchniowe występuje, gdy mamy do czynienia z fazą ciekłą, zaśdrugą fazą jest faza gazowa (powietrze, pary nasycone cieczy, inny gaz).W przypadku, gdy druga fazą jest faza skondensowana (inna ciecz nie mieszającasię z pierwszą lub ciało stałe) mówimy o napięciu międzyfazowym. Kształt,jaki przyjmuje kropla cieczy umieszczona na powierzchni ciała stałego zaleŜyod sił napięcia powierzchniowego i napięcia międzyfazowego. Definiuje teŜ ontzw. kąt zwilŜania :θθChem. Fiz. TCH II/23 10Dla ZPiPPK takŜe rozlewność.10
Napięcie powierzchniowe.MeniskNapięcie powierzchniowe powoduje tzw. wzniesienie kapilarne. Ciecz w rurcekapilarnej wznosi się (lub opada), dodatkowo tworząc menisk.meniskwklęsłyKształt menisku zależy odproporcji między napięciempowierzchniowym σ cg anapięciamimiędzyfazowymi σ sg i σ sc ,od czego zależy tez kątzwilżania θ.Można też powiedzieć, żezależy to od stosunku siłkohezji w cieczy (cc) iadhezji cieczy do ciałastałego (cs).meniskwypukłyChem. Fiz. TCH II/23 1111
Praca adhezji i praca kohezjiPraca potrzebna do rozerwania słupa cieczy o przekroju jednostkowym (zpominięciem pracy rozsunięcia fragmentów), czyli wyłącznie praca utworzenianowej (powiększenia istniejącej) powierzchni międzyfazowej wyniesie:Kohezja (spójność)substancji A:Adhezja (przyleganie)substancji A do B:AAAAZagadnienia te (adhezja,kohezja, zwilŜalność,rozlewność) odgrywająkluczową rolę wprzemyśle farb, lakierów iklejów.ABABwkohσChem. Fiz. TCH II/23 12= 2Awadh=A+B−ABσσσ12
Napięcie powierzchniowewybranych cieczyW tabeli podano wartości napięcia powierzchniowego, σ,wybranych cieczy w temperaturze 25 o C (lub 20 o C).cieczσmN/mcieczσmN/mwoda (p,pr) 71,97 chloroform 20 (p) 27,1rtęć (p,pr) 473,5 eter etylowy 20 (pr) 17,01aceton 20 (p,pr) 23,70 etanol 20 (p) 22,27benzen 20 (p) 28,87 etanol 20 (pr) 23,61CCl 420(p) 26,8 kwas masłowy 20 (p) 26,8Napięcie powierzchniowe mierzy się względem powietrza (p), pary nasyconejdanej cieczy (pr), mieszaniny obydwóch, lub innych gazów (H 2 , N 2 )Chem. Fiz. TCH II/23 14Napięcie powierzchniowe zawsze maleje ze wzrostem temperatury.Inne gazy stosuje przy pomiarach napięcia powierzchniowego cieczy takich jakstopione metale.Dla ZpPiPPk dodać o parachorze, regule Eotvosa i wzorze Ramsaya-Shieldsa&&&14
Krople, bańki, pęcherzyki.Zdefiniowaliśmy juŜ kroplę, jako oddzielny fragment fazy ciekłej, który w przypadkunieobecności sił zewnętrznych przyjmuje kształt kulisty.Przez bańkę rozumieć będziemy fazę gazową otoczona filmem (błonką) fazyciekłej. Jak zobaczymy, równieŜ i ona w przypadku nieobecności sił zewnętrznychprzyjmuje kształt kulisty.Przez pęcherzyk rozumieć będziemy fragment fazy gazowej we wnętrzu fazyciekłej. TakŜe i on w przypadku nieobecności sił zewnętrznych przyjmuje kształtkulisty, przy czym w rzeczywistości odkształca go nieco siła wyporu,powodująca unoszenie się pęcherzyków ku górze.Wszystkie wymienione tutaj twory i ich zachowanie w róŜnych warunkachodgrywają istotną rolę w technologii i inŜynierii chemicznej, zwłaszcza wprocesie flotacji.Chem. Fiz. TCH II/23 15Flotacja rozbudowana w ZPiPPK. &&&15
Ciśnienie w bańkach ipęcherzykach &&&Chem. Fiz. TCH II/23 1616
FlotacjaChem. Fiz. TCH II/23 17Tego w TCH nie będzie.17
PręŜność par cieczy nadpowierzchnią zakrzywionąChem. Fiz. TCH II/23 1818
Napięcie powierzchnioweroztworówRoztwory muszą wykazywać napięcie powierzchniowe zaleŜne od napięciapowierzchniowego składników, oraz składu. Tylko dla niektórych substancji jestto zaleŜność addytywna (dla roztworów doskonałych półempiryczny współczynnikβ=0):σ = x σ σ + β1 1+ x2 2x1x2Substancje rozpuszczone moŜna sklasyfikować następująco:1. Związki nieorganiczne w niewielkim stopniu wpływają na napięcie powierzchniowe,zazwyczaj nieco je podwyŜszając (wpływ twardości wody).2. Związki organiczne mają wpływ na napięcie powierzchniowe związkóworganicznych, w których się rozpuszczają, zgodnie z w/w wzorem.3. Związki organiczne rozpuszczalne w wodzie, zwłaszcza o charakterzerównocześnie hydrofilowym i hydrofobowym w znacznym stopniu zmniejszająnapięcie powierzchniowe wody. Są to związki powierzchniowoczynne (surfaktanty).Chem. Fiz. TCH II/23 1919
Napięcie powierzchnioweroztworów (2)Jednym z równań opisujących zaleŜność napięcia powierzchniowego roztworuod stęŜenia substancji powierzchniowo czynnej jest równanie Szyszkowskiego:σ = σ0− a ln(1 + bc)gdzie: σ – napięcie powierzchniowe roztworu o stęŜeniu c,σ 0 – napięcie powierzchniowe czystego rozpuszczalnika,a i b – stałe charakteryzujące substancję powierzchniowo czynną, przyczym stała a charakteryzuje rodzaj detergentu (szereg homologiczny,do którego on naleŜy), zaś stała b zaleŜy od liczby atomówwęgla w jego cząsteczce (pozycji w szeregu homologicznym).Równanie to bywa przedstawiane w postaci:gdzie:σ ⎛ c ⎞= 1 + Aln⎜1+ ⎟a 1 σ 0 ⎝ B ⎠A = ; B = σ bUwaga na jednostki!0Chem. Fiz. TCH II/23 2020
Izoterma GibbsaMuszą istnieć konkretne powody (fizykochemiczne podstawy), dla którychobecność substancji powierzchniowo czynnych (surfaktantów, detergentów)wpływa na napięcie powierzchniowe. W rzeczy samej, powodem jest fakt, Ŝecząsteczki detergentu gromadzą się w warstewce przypowierzchniowejroztworu.Nadmiar powierzchniowy, Γ (n) ( n)∆n⎡mol⎤2 , definiuje się jako: Γ2=⎢ 2A ⎣ m ⎥⎦przy czym: ∆n= ( c − c)⋅v= ( c − c)⋅δ⋅ Assgdzie: ∆n oznacza liczbę moli w warstewce przypowierzchniowej o powierzchniA i objętości v grubości δ,c s oznacza stęŜenie detergentu w warstewce przypowierzchniowej, zaśc oznacza stęŜenie detergentu w głębi roztworu (ang. bulk).Chem. Fiz. TCH II/23 2121
Izoterma Gibbsa (2)Gibbs udowodnił, Ŝe nadmiar powierzchniowy związany jest z napięciempowierzchniowym roztworu zgodnie z wzorem:( n)a ⎛ ∂ ⎞Γ2 = − ⎜ ⎟RTgdzie: a⎝ ∂aP Toznacza aktywność substancji rozpuszczonej,⎠ ,R – stałą gazową, T – temperaturę bezwzględną.zaś pochodna oznacza zaleŜność napięcia powierzchniowego od aktywności.Z dobrym przybliŜeniem (niskie stęŜenia) moŜna aktywność zastąpić stęŜeniemDla surfaktantów⎛ ∂σ⎞⎜ ⎟⎝ ∂c⎠P,T( n)c ⎛ ∂σ⎞Γ2 = − ⎜ ⎟RT ⎝ ∂c⎠< 0P,T, co oznacza, Ŝe( )Γ n2>czyli, Ŝe to one właśnie gromadzą się w warstewce przypowierzchniowej.Chem. Fiz. TCH II/23 220σ22
Izoterma Gibbsa (3)Maksymalny nadmiar powierzchniowy jest zdefiniowany jako:Γ=Γ( n)( )2maxlim n2c→∞i moŜna go obliczyć, jeŜeli pochodną do wzoru:Wyznaczymy np. z równania Szyszkowskiego:( n)c ⎛ ∂σ⎞Γ2 = − ⎜ ⎟RT ⎝ ∂c⎠⎛ ∂σ⎞ a ⋅b⎜ ⎟ = −⎝ ∂c⎠ 1+b ⋅c(gdzie wszystkie symbole mają znaczenie jak w tym równaniu)P,Ti otrzymamy:Γ( n)2max=1 ⎛ a ⋅b⋅clim⎜RT c →∞⎝1+bc⎞⎟⎠nΓ ( )2max=aRTChem. Fiz. TCH II/23 2323
Warstewki i miceleWyjaśnienia tendencji cząsteczek surfaktantu do samorzutnego gromadzeniasię przy powierzchni wody znajdziemy w ich strukturze. Znane są one takŜepod nazwą substancji amfifilowych, co wskazuje na obecność w ich strukturzezarówno fragmentów hydrofilowych, jak i hydrofobowych. Mają one najczęściejkształt wydłuŜony:fragmenthydrofobowycentrum (grupa)hydrofilowa, np. jonSposób gromadzenia się takich cząsteczek w warstwie przypowierzchniowejilustruje rysunek, na którym cząsteczki ustawiają się grupami hydrofilowymi downętrza roztworu.powierzchniaroztworu wodnegoChem. Fiz. TCH II/23 2424
Warstewki i micele (2)Maksymalny nadmiar powierzchniowy oznacza, Ŝe przy powierzchni więcejcząsteczek surfaktantu juŜ się nie zmieści. Zatem, moŜna wyliczyć tzw.powierzchnię siadania cząsteczki surfaktantu, ω, ze wzoru:ω= ( nΓ)2max1⋅ NWynik posiada wymiar m 2 (lub m 2 na cząsteczkę) i oznacza powierzchnięzajmowaną przez jedną cząsteczkę (w odpowiednim jej przekroju, z uwzględnieniemmoŜliwości ich upakowania .Avpowierzchniaroztworu wodnegoChem. Fiz. TCH II/23 25Dla ZPiPPK rozwinąć to znacznie. Nawet samoorganizujące się warstewkiLangmuira-Blodgett! &&&TakŜe o samych surfaktantach (detergenty, emulgatory, środki pianotwórcze).25
Warstewki i micele (3)Krzywa zaleŜności napięciapowierzchniowego odσstęŜenia ujawnia stęŜenie,powyŜej którego dalszedodawanie surfaktantu niec krma juŜ wpływu na σ (dalsze„modyfikowanie” powierzchni nie jest juŜ moŜliwe). Oznacza to osiągnięciecmaksymalnego nadmiaru powierzchniowego. Pytanie: co się dzieje z substancjąamfifilową, jeŜeli jednak nadal jej dodajemy do roztworu?Tworzą się wtedy aglomeraty podobne do przedstawionegoobok. Cząsteczki ustawiają się swoimi centrami hydrofilowymiw kierunku „na zewnątrz” (do roztworu wodnego), zaś centrumaglomeratu stanowią „splątane” łańcuchy hydrofobowe.Aglomerat taki nazywamy micelą, a stęŜenie zaznaczone nawykresie – krytycznym stęŜeniem micelizacji (KSM, CMC).Chem. Fiz. TCH II/23 26Uwaga o rozmieszczeniu ładunku w miceli. Dla ZPiPPK – gruntowniej. &&&26
AdsorpcjaAdsorpcja jest procesem samorzutnego gromadzenia się cząsteczek nagranicy faz (na powierzchni międzyfazowej). Cząsteczki te pochodzić mogątylko z tych faz, które mają mobilne cząsteczki (gaz lub ciecz). Substancjęgromadzącą się nazywamy adsorbatem zaś substancję stanowiącą fazę, naktórej powierzchni gromadzą się cząsteczki – adsorbentem. Cząsteczki adsorbentupochodzą z fazy przeciwnej do tej, na której powierzchni zachodzi adsorpcja.Jest to podstawa klasyfikacji adsorpcji wg rodzaju faz graniczących:adsorbentciało stałeciało stałecieczcieczfaza, z której pochodzi adsorbatgazowaciekłagazcieczW praktyce ograniczamy się do dwóch pierwszych układów. Ostatnie sytuacje, (ruchliwośćcząsteczek w obu fazach), są często jedynie etapami wstępnymi procesów znanychjako absorpcja i podział międzyfazowy.Chem. Fiz. TCH II/23 27W przypadku zjawiska opisywanego izotermą Gibbsa moŜna mówić o adsorpcji,choć trudno wskazać adsorbent (adsorbatem jest substancja powierzchniowoczynna). W kaŜdym razie gromadzenie się cząsteczek na granicy faz NIEJEST tu spowodowane„ niezrównowaŜonymi oddziaływaniami w fazieprzeciwnej do tej z której pochodzi adsorbat, choć – oczywiście – energiacząsteczek przy powierzchni jest inna od tych w głębi roztworu.27
Rodzaje adsorpcjiKryterium jest tu rodzaj oddziaływań adsorbent-adsorbat i tego konsekwencje:adsorpcja fizyczna (fizysorpcja)oddziaływania fizycznemałe ciepło adsorpcji (do 50 kJ/mol)moŜliwa adsorpcja wielowarstwowaenergia aktywacji niskaadsorpcja chemiczna (chemisorpcja)wiązania chemiczneduŜe ciepło adsopcji (kilkaset kJ/mol)adsorpcja jednowarstwowaenergia aktywacji wysokaW niektórych przypadkach (przy tym samym adsorbacie i adsorbencie, np. H 2na Ni) mechanizm adsorpcji moŜe ulegać zmianie w zaleŜności od temperatury– w niskich zachodzi fizysorpcja, a w wysokich – po przekroczeniu energiiaktywacji – pojawia się chemisorpcja.Chem. Fiz. TCH II/23 28Dla ZPiPPK sać bliŜsze info o tej zaleŜności temperaturowej. &&&28
Funkcje termodynamiczneadsorpcjiJeŜeli adsorpcja jest procesem samorzutnym, to entalpia swobodna adsorpcjimusi być zawsze ujemna: ∆G ads
Miary adsorpcjiPodstawową jednostką adsorpcji jest ilość adsorbatu przypadająca na jednostkępowierzchni adsorbentu: a [mol adsorbatu/m 2 adsorbentu]. Często wyraŜasię ją takŜe jako a’ [mol adsorbatu/ g adsorbentu]. Związek między a i a’ danyjest wzorem:a = a'⋅wgdzie w jest powierzchnią właściwą adsorbentu wyraŜoną w m 2 /g.MoŜliwe jest teŜ zdefiniowanie (jednoznacznie w przypadku adsorpcji jednowarstwowej)tzw. stopnia pokrycia θ powierzchni. Przy pełnym pokryciumonowarstwowym adsorpcja osiąga wartość maksymalną, a max . Zatem:θ =a =aa'amax' maxgdzie θ oznacza ułamek powierzchni adsorbentuzajętej przez adsorbat, zaś 1 – θ oznacza ułamekpowierzchni wolnej od adsorbatu. Maksymalnie θmoŜe oczywiście wynosić 1.Chem. Fiz. TCH II/23 30Spotyka się teŜ adsorpcję wyraŜana w jeszcze innych jednostkach (tzn. ilośćadsorbatu, np. w ml, czy cm 3 (oczywiście „normalnych”).30
Izotermy, izobary i izosteryadsorpcjiZaleŜność wielkości adsorpcji od ciśnienia cząstkowego (lub stęŜenia) adsorbatuw fazie z której zachodzi adsorpcja w warunkach stałej temperatury nazywamyizotermą adsorpcji. Będzie to zatem wykres w układzie współrzędnycha=f(P) a’=f(P) θ=f(P); T=const.ZaleŜność wielkości adsorpcji od temperatury w warunkach stałego ciśnieniacząstkowego (lub stęŜenia) adsorbatu w fazie z której zachodzi adsorpcja nazywamyizobarą adsorpcji. Będzie to zatem wykres w układzie współrzędnycha=f(P) a’=f(P) θ=f(P); P=const.ZaleŜność ciśnienia cząstkowego adsorbatu (lub stęŜenia) od temperatury wwarunkach stałej wielkości adsorpcji (ciśnienia niezbędnego do podtrzymaniastałej adsorpcji w warunkach zmiennej temperatury) nazywamy izosterą adsorpcji.Będzie to zatem wykres w układzie współrzędnychP=f(T); θ(a,a’)=const.Chem. Fiz. TCH II/23 3131
Izotermy, izobary i izosteryadsorpcji (2)Odpowiednie wykresy (przykładowe) wyglądają następująco:Izoterma adsorpcji:a=f(P) a’=f(P) θ=f(P); T=const.Izobara adsorpcji:a=f(P) a’=f(P) θ=f(P); P=const.aP 1P 2P 1>P 2>P 3aT 3>T 2>T 1PT 1T 2T 3Izostera adsorpcji:PP=f(T); θ(a,a’)=const.a 3P 3TlnP∆Hadsln P = − + constRTa 3a 1a2a 1>a 2>a 3a 1a 2TChem. Fiz. TCH II/23 321/TZlinearyzowana postać izostery przypomina (jest analogiczna do) równaniaClausiusa-Clapeyrona.32
ZałoŜenia Langmuira:Izoterma Langmuira1) na powierzchni adsorbentu znajdują się centra adsorpcyjne C, z którychkaŜde moŜe zaadsorbować jedną cząsteczkę adsorbatu A (wynika z tego,Ŝe maksymalna adsorpcja odpowiada monowarstwie), tworząc kompleksadsorpcyjny CA,2) wszystkie centra są równowaŜne pod względem energetycznym (energiitworzonych oddziaływań),3) nie dochodzi do ruchów zaadsorbowanych cząsteczek na powierzchniadsorbentu,4) oddziaływania między cząsteczkami adsorbatu w warstwie adsorpcyjnej sąpomijalne,5) w stanie równowagi szybkość adsorpcji i desorpcji jest taka sama,6) proces adsorpcji da się zapisać równaniem: C + A CAChem. Fiz. TCH II/23 3333
Izoterma Langmuira (2)C + ADla tak zapisanej reakcji moŜna zapisać jej stałą równowagi, przy czym miarą„aktywności” CA jest ułamek obsadzonych centrów adsorpcyjnych, czyli stopieńpokrycia powierzchni, θ, miarą „aktywności” C jest ułamek nie obsadzonychcentrów adsorpcyjnych, 1– θ, zaś miarą aktywności adsorbatu A, jego ciśnieniecząstkowe P. Stąd:θK =( 1−θ) PPo przekształceniu otrzymujemy równanie izotermy Langmuira, gdzie K nazywamystałą adsorpcji K ads ,KadsPθ = 1+ K Pktóra, zgodnie z rozwaŜaniami znanymi z kinetyki chemicznej, powinna byćrówna stosunkowi stałej szybkości adsorpcji do stałej szybkości desorpcji:kadsK =Chem. Fiz. TCH II/23 34kdesCAads34
Izoterma Langmuira (3)Wykres izotermy Langmuira przedstawia się następująco:KadsPθ = 1+ K Padsθθ=1Dla małych wartości P, gdy K ads P1, θ=1.θ=K adsPPCechy te: zakres proporcjonalności, jak i asymptotyczne dąŜenie stopnia pokryciado jedności, dobrze widać na wykresie.Gdy θ=1, adsorpcja a=a max (pełne obsadzenie monowarstwy).Izoterma Langmuira dobrze opisuje wiele układów, w szczególności chemisorpcjęz roztworów, kiedy to załoŜenie o monowarstwie jest dobrze spełnione.Chem. Fiz. TCH II/23 3535
Izoterma BETDla przypadku adsorpcji z fazy gazowej, gdziedoświadczalne zaleŜności często nie zgadzałysię z izotermą Langmuira, Brunauer, Emmet iTeller opracowali opis matematyczny adsorpcjiwielowarstwowej znany jako izoterma BET.Równanie to podaję tutaj bez wyprowadzenia.θ =⎛ ⎜1−⎝PP0CPP0⎞ ⎡⎟ ⋅ ⎢1+ ( C −1)⎠ ⎣P ⎤P⎥0 ⎦W równaniu tym:C =k'k Lgdzie: k’ to stała równowagi tworzenia pierwszej warstwy adsorpcyjnej,k L – stała równowagi kondensacji równa 1/P 0 ,P 0 – pręŜność pary nasyconej substancji adsorbowanej w danej temperaturzeChem. Fiz. TCH II/23 36Dla ZPiPPK oczywiście z wyprowadzeniem.Ten sam Teller, który jest bardziej znany jako fizyk jądrowy – współtwórcabomby atomowej i wodorowej.36
Izoterma BET (2)Równanie izotermy BET moŜna równieŜzapisać w postaci:a =⎛ ⎜1−⎝PP0amCPP0⎞ ⎡⎟ ⋅ ⎢1+ ( C −1)⎠ ⎣P ⎤P⎥0 ⎦W równaniu tym a m nie oznacza maksymalnej wartości adsorpcji, lecz jej wartośćodpowiadającą pojemności monowarstwy. Stąd wynika, Ŝe moŜliwa jestsytuacja, gdy:a > oraz θ >1a mBrunauer zaproponował klasyfikację adsorpcji wg typów obserwowanychizoterm adsorpcji i wyróŜnił 5 jej rodzajów.Chem. Fiz. TCH II/23 3737
θTyp IIzotermy adsorpcjiwielowarstwowej wg BrunaueraI – P 0 i C bardzo duŜe, równanie upraszczasię do izotermy Langmuira.θII – C przyjmuje wartości odkilku (3) do kilkusetTyp IIθTyp IIIIII – C≤1P/P 0Typy IV i V są modyfikacjami typówII i III. Fakt brkau wzrostu adsorpcjiwynika z faktu kondensacji kapilarnejw porach adsorbentu. Przy istnieniucieczy wzrost ciśnienia zewnętrznegonie prowadzi w danejtemperaturze do przekroczeniaciśnienia cząstkowego adsorbatupowyŜej P 0 .θTyp IVP/P 0θP/P 0Typ VChem. Fiz. TCH II/23 38P/P 0P/P 0=1P/P 0=1P/P 038
LepkośćLepkość, czyli współczynnik tarcia wewnętrznego płynu, η, definiuje nam wzórNewtona, opisujący przepływ laminarny (uwarstwiony):λvv+λ·dv/dxFdv= −ηdxAgdzie: F jest siłą tarcia między dwiema warstwami płynu o powierzchni styku A,przy gradiencie szybkości w kierunku normalnym do płaszczyzny warstwrównym dv/dx. Oś x jest tu osią pionową. Siła tarcia ma znak ujemny,bowiem skierowana jest przeciwnie do siły powodującej ruch (przepływ).Tak zdefiniowana lepkość jest znana jako lepkość dynamiczna.Chem. Fiz. TCH II/22 39Czy to jest wzór Newtona? Tak. Dla ZPiPPK dodać tiksotropy.39
Lepkość (2)Jednostkami lepkości dynamicznej są: F dx ⎡ N ⋅s⎤⎡ kg ⎤η = − ⋅⎢≡2A dv ⎣ m ⎥⎦⎢⎣m⋅s⎥⎦W starym systemie, były to puazy (poise), a praktycznie centypuazy, przy czym:N ⋅s1cP = 0,0012mJest to o tyle waŜne, Ŝe dane tablicowe są wciąŜ najczęściej wyraŜane w cP.Istnieje równieŜ wielkość zwana lepkością kinematyczną, ν, będąca stosunkiemlepkości dynamicznej do gęstości płynu:32η ⎡ N ⋅s⋅m⎤ ⎡m⎤ν = ⎢ ⎥ ≡2 ⎢ ⎥ρ ⎣ m ⋅kg⎦ ⎣ s ⎦Dawne jednostki to sztoksy (Stokes) i centysztoksy, przy czym:1cStokes = 10−6ms2Chem. Fiz. TCH II/22 40Ostatnie równanie zawiera gęstość, ρ, która jest równa m·N A /V m .40
Lepkość (3)Metody pomiaru – wiskozymetr Ubbelohdego:Wzór Hagena-Poiseulle’a:4πr∆Pv = τ8lηChem. Fiz. TCH II/22 41Równanie (2) jest równaniem Poiseuille’a, gdzie r jest promieniem rurkikapilarnej, l – jej długością, ∆P – róŜnicą ciśnień między wylotem a wlotem, τ -czasem wypływu, zaś v – objętością gazu jaka przepłynęła przez rurkę.Ostatnie równanie zawiera gęstość, ρ, która jest równa m·N A /V m .41
Lepkość (4)Lepkość roztworów. Związek lepkości z masą molową.ηη =wzgl [ η]η 0ηwzgl−1= limc →0 cRównanie Marka- Houwinka. :a[ η] = K ⋅ MLepkość koloidów, równanie Einsteina. :( 1+2, Φ)η = 5η 0Chem. Fiz. TCH II/22 4242
Prawo StokesaStokes wykazał, Ŝe współczynnik oporu tarcia stawiany przez ośrodek (płyn)o lepkości η ciału o kształcie kuli o promieniu r dany jest wzorem: f = 6π ⋅r⋅ηa siła oporu tarcia zaleŜy od szybkości ruchu cząstki, v;WF tG= f ⋅v= 6π ⋅r⋅η⋅vJeŜeli kulista cząstka o gęstości ρ i promieniu r opada ruchem jednostajnymw ośrodku o lepkości η i gęstości ρ 0 ,to oznacza, Ŝe siłacięŜkości G jest zrównowaŜona siłą wyporu W i siłą oporów tarcia F t :4 34 3G Ft + W; π ⋅r⋅ ρ ⋅ g = 6π⋅r⋅η⋅v+ π ⋅r⋅ ρ ⋅ g=3304 34 33π ⋅r⋅ ρ ⋅ g −3π ⋅r⋅ ρ0⋅ g = 6π⋅r⋅η⋅v2 2r ( )9⋅ ρ − ρ0⋅ g = η ⋅vOstatni wzór pozwala na obliczenie szybkości opadania przy znanych parametrachkulki i ośrodka lub lepkości, przy znanych parametrach kulki, ośrodka izmierzonej szybkości opadania (wiskozymetr Höplera).Chem. Fiz. TCH II/24 43F tCząstka zaczyna opadać ruchem przyspieszonym o przyśpieszeniu malejącym wczasie, bowiem siła oporów tarcia rośnie ze wzrostem prędkości opadania.43
Separacja hydraulicznaOstanie równania moŜna teŜ zapisać w postaci:ρ0G = Ft + W;m⋅g = 6π ⋅r⋅η⋅v+ m⋅⋅ gρ⎛ ρ ⎞ ⎛ ⎞m ⋅ ⎜ −0ρg m ⋅ ⎜ −01 ⎟⋅ 1 ⎟⋅ gdhv⎝ ρ ⎠=⎝ ρ ⎠i przekształcić do postaci: = == S ⋅ gdt 6π⋅r⋅ηf⎛ ρ ⎞m ⋅ ⎜ 1−0⎟Gdzie wyraŜenie S nosi nazwę współczynnika sedymentacji S =⎝ ρ ⎠fW oparciu o te równania moŜna obliczyć czas potrzebny do opadnięcia ookreśloną wysokość h cząstki o danej masie i rozmiarach w konkretnymośrodku.Chem. Fiz. TCH II/24 4444
Separacja hydrauliczna (2)W praktyce omawiane zjawiska wykorzystuje się w urządzeniach do segregacjizawiesin wg masy (wielkości) cząstek fazy rozproszonej na drodze tzw.separacji hydraulicznej. Uproszczony schemat urządzenia pokazany jestponiŜej:kierunek ruchu (przepływu) zawiesinyprzelew wylotowy(odpływowy)przelew wlotowyhleje odbiorczenajwiększe cząstkinajdrobniejsze cząstkiChem. Fiz. TCH II/24 45Tak teŜ separuje się pył wulkaniczny w powietrzu, największe (najcięŜsze cząstkiopadają najbliŜej krateru niesione wiatrem.Tory cząstek nie do końca odpowiadają rzeczywistym.45
Separacja hydrauliczna (3)W przypadku bardzo drobnych cząstek (koloidalnych), które podlegają prawudyfuzji, segregacji w kierunku w dół przeciwstawia się dyfuzja, skierowana wkierunku przeciwnym do powstającego gradientu stęŜenia, zatem w górę.Wtedy moŜe nie dojść do opadania cząstek, lecz moŜe ustalić się gradientstęŜeń (u dołu wyŜsze).W stanie równowagi:Przy czym współczynnik dyfuzji dany jest kB ⋅TD =równaniem Stokesa-Einsteina:fRozwiązanie równania róŜniczkowego dajewzór określający gradient stęŜenia wzdłuŜosi pionowej (wysokości – h):clnc21dhdtc − D⎛ ρ0⎞m ⋅ ⎜1− ⎟g⎝ ρ=⎠k TBdcdh= 0( h − h )21Chem. Fiz. TCH II/24 46k B jest oczywiście stałą Boltzmanna.Dla ZPiPPK – całkowanie. &&&46
Separacja hydrauliczna w polusiły odśrodkowejO równowadze sedymentacyjnej decyduje wartość przyspieszenia. W polugrawitacyjnym powstający gradient moŜe być bardzo mały w przypadku bardzomałych cząstek. MoŜna to przezwycięŜyć uŜywając wirówek. Wtedy szybkośćprzesuwania się cząstek (radialna – wzdłuŜ promienia r).W stanie równowagi:dr dcc − D = 0dt drRozwiązanie równania róŜniczkowego dajewzór określający gradient stęŜenia wzdłuŜpromienia r):clncGdzie szybkość kątowa ω zaleŜy od liczby obrotów na minutę n:21dr2v = = S ⋅ω rdt⎛ ρ0⎞ 2m ⋅ ⎜1− ⎟ω⎝ ρ ⎠ 2 2=( r2− r1)2kTChem. Fiz. TCH II/24 47B2πnω =60Metody te uŜywane są do frakcjonowania białek, a nawet izotopów. Niedawnoopracowano takŜe metodę frakcjonowania cząstek zawiesin w rotującychkolumnach „chromatograficznych”, znaną jako FFF (Field Flow Fractionation).Stąd problem irańskich wirówek.FFF – patrz „Nowe Horyzonty …”, CEEAM, Wydawnictwo PG, Gdańsk 2003,str.417-444.Dla ZPiPPK – całkowanie. &&&47