

Glykogenose
Glykogenose
Glykogenose

Sie wollen auch ein ePaper? Erhöhen Sie die Reichweite Ihrer Titel.
YUMPU macht aus Druck-PDFs automatisch weboptimierte ePaper, die Google liebt.
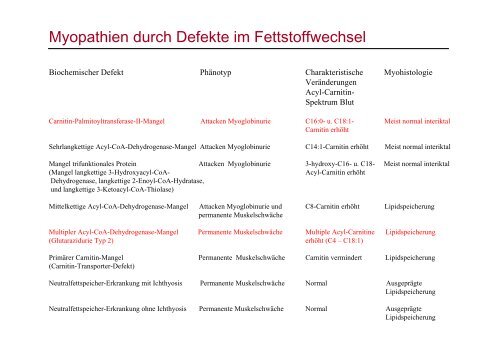
Myopathien durch Defekte im Fettstoffwechsel<br />
Biochemischer Defekt Phänotyp Charakteristische Myohistologie<br />
Veränderungen<br />
Acyl-Carnitin-<br />
Spektrum Blut<br />
Carnitin-Palmitoyltransferase-II-Mangel Attacken Myoglobinurie C16:0- u. C18:1- Meist normal interiktal<br />
Carnitin erhöht<br />
Sehrlangkettige Acyl-CoA-Dehydrogenase-Mangel Attacken Myoglobinurie C14:1-Carnitin erhöht Meist normal interiktal<br />
Mangel trifunktionales Protein Attacken Myoglobinurie 3-hydroxy-C16- u. C18- Meist normal interiktal<br />
(Mangel langkettige 3-Hydroxyacyl-CoA- Acyl-Carnitin erhöht<br />
Dehydrogenase, langkettige 2-Enoyl-CoA-Hydratase,<br />
und langkettige 3-Ketoacyl-CoA-Thiolase)<br />
Mittelkettige Acyl-CoA-Dehydrogenase-Mangel Attacken Myoglobinurie und C8-Carnitin erhöht Lipidspeicherung<br />
permanente Muskelschwäche<br />
Multipler Acyl-CoA-Dehydrogenase-Mangel Permanente Muskelschwäche Multiple Acyl-Carnitine Lipidspeicherung<br />
(Glutarazidurie Typ 2) erhöht (C4 – C18:1)<br />
Primärer Carnitin-Mangel Permanente Muskelschwäche Carnitin vermindert Lipidspeicherung<br />
(Carnitin-Transporter-Defekt)<br />
Neutralfettspeicher-Erkrankung mit Ichthyosis Permanente Muskelschwäche Normal Ausgeprägte<br />
Lipidspeicherung<br />
Neutralfettspeicher-Erkrankung ohne Ichthyosis Permanente Muskelschwäche Normal Ausgeprägte<br />
Lipidspeicherung














