Ph.D. THESIS ABSTRACT SYNTHESIS, STEREOCHEMISTRY AND ...
Ph.D. THESIS ABSTRACT SYNTHESIS, STEREOCHEMISTRY AND ...
Ph.D. THESIS ABSTRACT SYNTHESIS, STEREOCHEMISTRY AND ...

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
“BABES-BOLYAI” UNIVERSITY CLUJ-NAPOCA<br />
FACULTY OF CHEMISTRY <strong>AND</strong> CHEMICAL ENGINEERING<br />
ORGANIC CHEMISTRY DEPARTMENT<br />
<strong>Ph</strong>.D. <strong>THESIS</strong> <strong>ABSTRACT</strong><br />
SYN<strong>THESIS</strong>, <strong>STEREOCHEMISTRY</strong> <strong>AND</strong> PROPERTIES OF SOME NEW<br />
SPIRANE-TYPE COMPOUNDS CONTAINING 1,3-DIOXANE UNITS,<br />
<strong>AND</strong> OF SOME NEW MACROCYCLES WITH PHENOTHIAZINE UNITS<br />
<strong>Ph</strong>.D. STUDENT SCIENTIFIC ADVISER<br />
RADU GROPEANU Prof. Univ. Dr. ION GROSU<br />
JURY<br />
PRESIDENT<br />
Prof. Dr. CATALIN POPESCU – Head of the Chemistry-<strong>Ph</strong>ysics Department, Faculty of<br />
Chemistry and Chemical Engineering, Cluj-Napoca<br />
REVIEWERS<br />
Prof. Dr. THOMAS J.J. MÜLLER– Karl Ruprecht University, Heidelberg (Germany)<br />
Prof. Dr. IONEL MANGALAGIU – Al. I. Cuza University, Iasi<br />
Prof. Dr. SORIN MAGER – Babeş-Bolyai University, Cluj-Napoca<br />
Defense: November 10 th<br />
-2005-
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
CONTENTS:<br />
PART A.<br />
SYN<strong>THESIS</strong>, <strong>STEREOCHEMISTRY</strong> <strong>AND</strong> PROPERTIES OF SOME NEW SPIRANE-<br />
TYPE COMPOUNDS CONTAINING 1,3-DIOXANE UNITS<br />
1. SYN<strong>THESIS</strong> <strong>AND</strong> STREOCHEMICAL STUDY OF SOME NEW BENZO-SPIRANES<br />
CONTAINING 1,3-DIOXANE UNITS<br />
1.1. GENERAL ASPECTS CONCERNING THE <strong>STEREOCHEMISTRY</strong> OF 1,3-DIOXANE<br />
SPIRANES<br />
1.1.1. GENERALITIES<br />
1.1.2. THERMODINAMIC <strong>AND</strong> KINETIC ASPECTS OF 1,3-DIOXANE RING<br />
1.1.3. FLEXIBLE <strong>AND</strong> ANANCOMERIC 1,3-DIOXANE COMPOUNDS<br />
1.1.4. CONFORMATIONAL ANALYSIS OF 2-ARYL-1,3-DIOXANE DERIVATIVES<br />
1.1.5. <strong>STEREOCHEMISTRY</strong> OF SPIRO-COMPOUNDS WITH 6-MEMBERED RINGS<br />
1.2. <strong>STEREOCHEMISTRY</strong> OF BENZO-1,3-DIOXANES<br />
1.2.1. <strong>STEREOCHEMISTRY</strong> OF CYCLOHEXENE<br />
1.2.2. BENZO-1,3-DIOXANE<br />
1.3. ORIGINAL CONTRIBUTIONS<br />
1.3.1. INTRODUCTION<br />
1.3.2. SYN<strong>THESIS</strong> <strong>AND</strong> STRUCTURAL ASPECTS IN SOLUTION<br />
1.3.3. STEREOCHEMICAL ASPECTS OF BENZO-SPIRANES IN SOLID STATE<br />
2. SYN<strong>THESIS</strong>, <strong>STEREOCHEMISTRY</strong> <strong>AND</strong> ADSORPTION STUDIES OF SOME NEW<br />
(POLY)SPIRANES CONTAINING 1,2-DITHIOLANE UNITS<br />
2.1. GENERAL ASPECTS OF 1,2-DITHIOLANE<br />
2.1.1. MAIN METHODS OF SYN<strong>THESIS</strong><br />
2.1.2. <strong>STEREOCHEMISTRY</strong> OF THE 1,2-DITHIOLANE CYCLES<br />
2.1.3. ADSORPTION PROPERTIES OF ORGANOSULPHUR COMPOUNDS<br />
2.2. ORIGINAL RESULTS<br />
2.2.1. INTRODUCTION<br />
2.2.2. SYN<strong>THESIS</strong> <strong>AND</strong> <strong>STEREOCHEMISTRY</strong> OF SPIRANES CONTAINING<br />
MARGINAL 1,2-DITHIOLANE UNITS<br />
2.2.3. ADSORPTION STUDIES<br />
3. CONCLUSIONS<br />
4. EXPERIMENTAL<br />
4.1. GENERALITIES<br />
4.2. SYN<strong>THESIS</strong> <strong>AND</strong> DESCRIPTION OF THE COMPOUNDS<br />
REFERENCES<br />
LIST OF THE SYNTHESIZED COMPOUNDS<br />
PART B.<br />
SYN<strong>THESIS</strong> <strong>AND</strong> PHYSICO-CHEMICAL PROPERTIES OF SOME NEW<br />
MACROCYCLES CONTAINING 10H-PHENOTHIAZINE UNITS<br />
1. INTRODUCTION<br />
2. ORIGINAL RESULTS<br />
2.1. IODO-SUBSTITUTED PHENOTHIAZINES<br />
2.1.1. LITERATURE DATA ON THE IODO-PHENOTHIAZINES<br />
2.1.2. SYN<strong>THESIS</strong> OF IODO-PHENOTHIAZINE DERIVATIVES<br />
2.1.3. PALLADIUM-CATALYZED CROSS-COUPLING REACTIONS OF HALO-<br />
PHENOTHIAZINES<br />
2.2. FERROCENO-PHENOTHIAZINE MACROCYCLES<br />
2.2.1. SYN<strong>THESIS</strong> OF FERROCENE-PHENOTHIAZINE DERIVATIVES<br />
2
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
2.2.3. SYN<strong>THESIS</strong> OF MACROCYCLES CONTAINING FERROCENE <strong>AND</strong><br />
PHENOTHIAZINE UNITS<br />
2.2.4. PHYSICO-CHEMICAL PROPERTIES OF THE SYNTHESIZED DYADS <strong>AND</strong><br />
TRIADS<br />
2.3. SHAPE-PERSISTENT MACROCYCLES<br />
2.3.1. INTRODUCTION<br />
2.3.2. THE STRATEGY OF THE PHENOTHIAZINE-ACETYLENE MACROCYCLES<br />
SYN<strong>THESIS</strong><br />
2.3.3. THE SYN<strong>THESIS</strong> OF THE PRECURSORS<br />
2.3.4. THE CYCLIZATION ATTEMPT<br />
2.3. PHENOTHIAZINE CORON<strong>AND</strong>S<br />
2.3.1. GENERALITIES<br />
2.3.2. SYN<strong>THESIS</strong> OF MACROCYCLES STARTING FROM DIPHENOL<br />
DERIVATIVES<br />
2.3.3. THE SYN<strong>THESIS</strong> OF THE INTERMEDIATES<br />
2.3.4. SYN<strong>THESIS</strong> OF CORON<strong>AND</strong>S HAVING PHENOTHIAZINE UNITS<br />
3. CONCLUSIONS OF PART B<br />
4. EXPERIMENTAL<br />
REFERENCES<br />
LIST OF THE SYNTHESIZED COMPOUNDS<br />
KEYWORDS:<br />
1,3-dioxane; benzo-1,3-dioxane; (poly)spirane; benzo-spirane; 1,2-dithiolane; cyclic disulfide;<br />
iodo-phenothiazine; oligophenothiazine; ferrocene; Suzuki cross-coupling; Negishi crosscoupling;<br />
Sonogashira cross-coupling; McMurry reductive coupling; cyclophane; coronand;<br />
stereochemical analysis; adsorption on gold; adsorption on gold nanoparticles.<br />
INTRODUCTION<br />
The research work presented in this <strong>Ph</strong>.D. Thesis is conducted in two main fields<br />
represented one side by the study on the synthesis, stereochemistry and properties of some new<br />
derivatives of saturated and unsaturated heterocycle spiranes, and on the other side by the<br />
synthesis and properties of new phenothiazine oligomers and macrocycles containing<br />
phenothiazine units.<br />
The first subject of the thesis was developed at “Babes-Bolyai” University Cluj-Napoca<br />
in the research group I am belonging, while the investigations concerning the second subject of<br />
my thesis were made in the research group of Prof. Thomas Müller (Ruprecht Karls Universität<br />
Heidelberg) during a 12 months research stage financed by a DAAD fellowship (iodophenothiazines,<br />
oligophenothiazenes and ferrocene-phenothiazine macrocycles), and in Cluj-<br />
Napoca (phenothiazine coronands).<br />
The research concerning the saturated six-membered ring heterocycles was first oriented<br />
to the study of the stereochemistry of benzo-spiranes by means of NMR and X-ray diffraction.<br />
Another direction of the research was represented by the synthesis of some new spiranes<br />
containing 1,2-dithiolane units as cyclic disulfides, and the adsorption of these on the gold<br />
surface in order to obtain 2D and 3D monolayers of (poli)spiranes.<br />
The objectives of the research on phenothiazines were dedicated to the synthesis of<br />
oligophenothiazines as well as ferrocene-phenothiazine derivatives, including ferrocenephenothiazine<br />
macrocycles. Also, the obtainment of phenothiazine coronands as possible<br />
selective cation receptors was investigated and realized.<br />
3
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
PART A.<br />
SYN<strong>THESIS</strong>, <strong>STEREOCHEMISTRY</strong> <strong>AND</strong> PROPERTIES OF SOME NEW<br />
SPIRANE-TYPE COMPOUNDS CONTAINING 1,3-DIOXANE UNITS<br />
1.1. INTRODUCTION<br />
1,3-Benzodioxanes represent an interesting motif in applied chemistry. They were reported as<br />
agrochechemical fungicides, 1 biocides, 2 pesticides 3 and herbicides. 4 Some 1,3-benzodioxanes<br />
were tested for pharmacological activity and were found to have antiinflammatory activity and<br />
low toxicity, 5 or antiinflammatory, analgesic, mucolytic, and antipiretic activity. 6 The<br />
unsaturated spiranes occur in the acidic degradation of steroides 7 or are a part of the skeleton of<br />
saponine of Ruscus aculeatus L. 8 Despite the various and numerous reports, few papers are<br />
dealing with the stereochemistry of 1,3-benzodioxanes. 9 This prompted us to investigate in<br />
details the stereochemistry of a series of benzocondensed dioxa-monospiranes and of a<br />
dibenzocondensed tetraoxa-dispirane.<br />
1.2. SYN<strong>THESIS</strong> <strong>AND</strong> STRUCTURAL ASPECTS IN SOLUTION<br />
The synthesis of one of the target compounds (2) was previously described. 10 We have<br />
obtained the benzo-spiranes 11 by ketalization of several cyclohexanones with salicylic alcohol in<br />
fair to good yields (Scheme 1).<br />
4<br />
1<br />
1<br />
OH<br />
OH<br />
OH<br />
Benzene, reflux<br />
OH<br />
2 O O<br />
APTS<br />
- 4 H2O<br />
O<br />
R<br />
Benzen, reflux<br />
APTS<br />
- H2O<br />
R Product Yield<br />
H 2 40<br />
8-CH3 3 38<br />
8-R(+)-CH3 4 42<br />
9-CH3 5 40<br />
9-C(CH3)3 6 40<br />
5<br />
7 O<br />
4<br />
3<br />
10<br />
8 6<br />
11 O 9 O<br />
1<br />
16<br />
O 15<br />
12 14<br />
13<br />
2<br />
7<br />
Scheme 1<br />
10<br />
2<br />
11<br />
1<br />
O<br />
6<br />
3<br />
4<br />
2-6<br />
7<br />
O<br />
5<br />
+<br />
9<br />
R<br />
8<br />
10<br />
8<br />
7<br />
4<br />
5<br />
O<br />
6<br />
3<br />
11 O 9 O<br />
1<br />
2<br />
12<br />
16<br />
O 15<br />
14<br />
13<br />
The reactions equilibra were shifted toward products by using anhydrous sodium sulfate<br />
to remove the reaction water. The flash chromatography (silica gel, petroleum ether:ethyl<br />
acetate=10:1) was used to purify the crude products.<br />
The 1 H-NMR spectra of the products present the signals corresponding to the<br />
cyclohexane ring (δ = 1-2.15 ppm), which are slightly shifted to higher fields than the<br />
1<br />
Simon,W.E.J. Can. Pat. Appl. CA 2117503 AA, 1995.<br />
2<br />
Simon, W.E.J. Eur. Pat. Appl. EP 645373 A1, 1995.<br />
3<br />
a) Anderson, M.; Brinnand, A.G.; Woodall, R.E. Eur. Pat. Appl. EP 525877 A1, 1993. b) Anderson, M.; Brinnand,<br />
A.G.; Woodall, R.E. Eur. Pat. Appl. EP 469686 A1, 1992.<br />
4<br />
Enomoto, M.; Nagano, H.; Haga, T.; Morita, K.; Sato, M. Jpn. Kokai Tokkyo Koho JP 63275580 A2, 1988.<br />
5<br />
Dauksas, V.; Gaidelis, P.; Brukstus, A.; Ramanauskas, J.; Labanauskas, L.; Gasperaviciene, G.; Jautakiene, I.;<br />
Lapinskas, V.; Lauzikiene, N. Khim-Farm. Zh. 1989, 23(8), 942.<br />
6<br />
Torre, A. Eur Pat Appl EP 272223 A1, 1988.<br />
7<br />
a) Koga, T.; Nogami, Y. Tetrahedron Lett. 1986, 27, 4505. b) Izawa, H.; Katada, Y.; Sakamoto, Y.; Sato, Y.<br />
Tetrahedron Lett. 1969, 10, 2947.<br />
8<br />
Lapin, H.; Sannie, C. Bull. Soc. Chim. Fr. 1955, 1552.<br />
9<br />
Brink, M. Monatsh. Chem. 1973, 104, 619.<br />
10<br />
Chondhury, P.K.; Almene, J.; Foubelo, F.; Yus, M. Tetrahedron 1997, 53, 17373.<br />
11<br />
Gropeanu, R.A.; Grosu, I. “SYN<strong>THESIS</strong> <strong>AND</strong> <strong>STEREOCHEMISTRY</strong> OF SOME NEW SPIRO BENZO-1,3-<br />
DIOXANE DERIVATIVES” Central European Journal of Chemistry 2005, submitted.<br />
8<br />
4
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
corresponding signals of the starting cyclohexanones. Beside of these, the spectra present the<br />
aromatic protons (δ = 6.70-7.20 ppm) and the 1,3-dioxine methylene (δ = 4.80-4.90 ppm).<br />
Monospirane 2 has a flexible structure, both 4[H]-1,3-dioxine (B) and cyclohexane (A)<br />
rings are flipping (Scheme 2). The 4[H]-1,3-dioxine prefers the chiral half-chair conformations<br />
with C6 and O5 out of the plain of the aromatic ring. The flipping of the heterocycle determines<br />
an enantiomeric inversion (I and III; II and IV are enantiomers). The flipping of the cyclohexane<br />
ring represents diastereomeric equilibrium, structures I and III exhibit the O-C6H4- moiety in<br />
equatorial orientation while structures II and IV have the O-CH2-C6H4- group in equatorial<br />
position (refereed to the cyclohexane ring).<br />
I<br />
O<br />
B<br />
O<br />
O<br />
B<br />
O<br />
A<br />
A<br />
1 (A)<br />
2 (B) 4 (B)<br />
3 (A)<br />
O<br />
B<br />
O<br />
O<br />
B<br />
O<br />
III IV<br />
2<br />
Scheme 2<br />
These possibilities have as result a fast equilibrium between the four conformers at room<br />
temperature in solution, which was shown by the 1 H-NMR spectrum. The 1,3-dioxine methylene<br />
(position 4) gives rise to a singlet at δ 4.81 ppm, and the axial and equatorial cyclohexane<br />
protons could not been distinguished, the corresponding signals being mediated.<br />
Compounds 3-6 have semiflexible structures, the substituted cyclohexane ring is<br />
anancomeric (the R group is holding group and exhibits equatorial orientation) and the 4[H]-1,3dioxine<br />
ring is flipping (Schemes 3). These compounds show cis (scheme 16: VIa and VIb;<br />
scheme 17: VII and VIII) and trans isomers (scheme 3: Va and Vb; scheme 4: IX and X). R and<br />
O-C6H4- are considered as references. At the same time the chirality (planar) of the 4[H]-1,3dioxine<br />
ring determines the chirality of the compounds and the flipping of the heterocycle<br />
determines the enantiomeric inversion for 3-5 (Va Vb; VIa VIb) and the diastereomeric<br />
equilibrium for 6 (VII VIII; IX X). The cis and trans structures could not be isolated as<br />
single isomers and the compounds were investigated as mixture of isomers.<br />
Va<br />
O<br />
B<br />
O<br />
O<br />
B<br />
O<br />
A<br />
R<br />
A<br />
O<br />
A<br />
B<br />
pR B pS<br />
O<br />
R<br />
R<br />
trans<br />
Vb<br />
B<br />
VIa pS cis<br />
5,6<br />
VIb pR<br />
Scheme 3<br />
O<br />
A Me<br />
O<br />
A Me<br />
B<br />
*<br />
O<br />
(R) B<br />
B (R) *<br />
O<br />
pR pS<br />
VII cis<br />
VIII<br />
(R) *<br />
O<br />
B<br />
O<br />
A<br />
Me<br />
B<br />
O<br />
B<br />
O<br />
II<br />
*<br />
(R)<br />
IX pS trans<br />
4<br />
X pR<br />
Scheme 4<br />
O<br />
B<br />
O<br />
A<br />
Me<br />
A<br />
A<br />
A<br />
R<br />
5
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
The 1 H-NMR spectra of 3-6 show many signals in the part of the spectrum corresponding<br />
to the protons of the cyclohexane ring due to frozen flipping of this ring and to the differentiation<br />
of axial and equatorial orientations. The singlet corresponding to the CH2 protons of the<br />
heterocycle is more shielded for the trans isomer (∆δ 3-6 = δ cis - δ trans = 0.05-0.10 ppm).<br />
The diastereoisomers have only very small differences of the physico-chemical<br />
properties, so it was difficult to separate them and duly characterize. After the column<br />
chromatography separation, only samples enriched in one isomer could been obtained. The ratio<br />
between isomers was determined comparing the 1,3-dioxine methylene signals and the results<br />
are presented in Table 1.<br />
Table 1. Ratio of diastereomers in enriched fractions and the chemical shift of the corresponding<br />
1,3-dioxine methylene (CDCl3, 300 MHz, rt)<br />
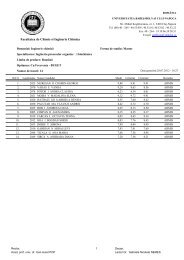
Entry Compound<br />
Ratio of diastereomers (DIA 1:DIA 2)*<br />
First sample Last sample<br />
Chemical shift (δ, ppm)<br />
DIA 1 DIA 2<br />
1 3 82:18 27:73 4.80 4.89<br />
2 4 80:20 34:66 4.80 4.89<br />
3 5 100:0 24:76 4.80 4.87<br />
4 6 58:42 42:58 4.80 4.89<br />
* The two isomers were formally named DIA 1 and DIA 2 tacking into account the Rf values.<br />
The exception was brought by the dispirane obtained by ketalization of 1,4-cyclohexanedione,<br />
which presents two well distinguished and separable isomers: 7 (trans) and 8 (cis).<br />
Dibenzo-dispiro derivatives 7 and 8 exhibit three stereogene elements, two 4[H]-1,3dioxine<br />
rings with planar chirality 12 and the differently substituted cyclohexane ring which<br />
determines cis and trans isomers. Cis and trans isomers of 6 were separated by flash<br />
chromatography and were investigated as single compounds. The chirality of the heterocycles<br />
determines like (pRpR; pSpS) and unlike (pRpS = pSpR) structures (Schemes 5 and 6). Cis<br />
isomer (XI-XIII) exhibits the reference groups (–C6H4-O-) in axial-equatorial orientations and<br />
the flipping of the cyclohexane ring is an homomeric equilibrium while the flipping of 4[H]-1,3dioxine<br />
rings determines the diastereoisomeric equilibrium. Trans isomer (XIV-XIX) exhibits<br />
the reference groups either in axial-axial, either in equatorial-equatorial orientations. The<br />
flipping of the cyclohexane or of the 4[H]-1,3-dioxine rings represents diastereoisomeric<br />
equilibria.<br />
B<br />
O<br />
O<br />
B<br />
O<br />
O<br />
O<br />
A<br />
B'<br />
O<br />
B<br />
O<br />
A<br />
O<br />
B'<br />
O<br />
B'<br />
O<br />
A<br />
O<br />
B'<br />
O<br />
pRpR<br />
B<br />
O<br />
pSpR<br />
B<br />
O<br />
pSpS<br />
XI XII<br />
XIII<br />
A A<br />
A<br />
A<br />
XI<br />
O<br />
B'<br />
O<br />
pRpR<br />
B<br />
O<br />
B<br />
O<br />
XII<br />
B'<br />
A O<br />
B'<br />
O<br />
8<br />
Scheme 5<br />
pSpR<br />
O<br />
B<br />
O<br />
A O<br />
B'<br />
O<br />
XIII pSpS<br />
12 Eliel, E.L.; Wilen, S.H. Configuration and Conformation of Cyclic Molecules. In: Stereochemistry of Organic<br />
Compounds (1994), John Wiley and Sons, Inc., New York, p.726.<br />
6
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
Cis and trans isomers are flexible compounds and the flipping of the central cyclohexane ring<br />
and of the heterocycles renders in equilibrium all possible isomers (enantiomers and<br />
diastereoisomers; cis: XI-XII, trans: XIV-XIX). The rt 1 H NMR spectra of trans and cis exhibit<br />
only singlets for the protons of 4[H]-1,3-dioxine rings (7-trans: δ4,13 = 4.85 ppm; 7-cis: δ4,11 =<br />
4.86 ppm). The spectra display a singlet for the protons of the cyclohexane ring of the trans<br />
isomer and overlapped multiplets for those of cis one (7: δ7,8,15,16 = 2.03 ppm; 8: δ7,8,15,16 = 1.93-<br />
2.09 ppm; Schemes 5, 6).<br />
B<br />
O<br />
B<br />
O<br />
O<br />
O<br />
XIX (aa)<br />
O<br />
A<br />
B'<br />
O B<br />
O<br />
A<br />
O<br />
B'<br />
O<br />
B'<br />
O<br />
A<br />
O<br />
B'<br />
O<br />
pRpR<br />
XIV (ee)<br />
B<br />
O pSpR<br />
XV (ee)<br />
B<br />
O<br />
XVI (ee)<br />
pSpS<br />
A<br />
A O<br />
B'<br />
pRpR<br />
O<br />
B<br />
B<br />
O<br />
O<br />
A<br />
A O<br />
B'<br />
O<br />
pSpR<br />
XVIII (aa)<br />
7<br />
Scheme 6<br />
B'<br />
B<br />
O<br />
O<br />
XVII (aa)<br />
A<br />
A O<br />
B'<br />
O<br />
pSpS<br />
The variable temperature 1 H NMR experiments run with 7 and 8 (Figure 1 and 2) show<br />
the freezing of the flipping of the rings at low temperatures (that determines a higher number of<br />
signals in the NMR spectra). Two net coalescence points for 7 isomer (Tc = 224 K for the signals<br />
belonging to the protons of the cyclohexane ring, Tc’ = 220 K for the protons of the heterocycles)<br />
and one coalescence point for cis isomer [the signals corresponding to the 4[H]-1,3-dioxine ring<br />
(Tc = 217 K)] could be measured.<br />
5.2<br />
5.0<br />
4.8<br />
4.6<br />
4.4<br />
4.2<br />
4.0<br />
Figure 1. 1 H-NMR spectra of 7 recorded at different temperatures (CDCl3, 300 MHz)<br />
3.8<br />
O<br />
O<br />
3.6<br />
3.4<br />
O<br />
293 K<br />
233 K<br />
225 K<br />
224 K<br />
223 K<br />
220 K<br />
217 K<br />
213 K<br />
O<br />
3.2<br />
3.0<br />
2.8<br />
2.6<br />
2.4<br />
2.2<br />
2.0<br />
1.8<br />
1.6<br />
7
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
5.2<br />
5.0<br />
4.8<br />
4.6<br />
4.4<br />
4.2<br />
4.0<br />
3.8<br />
3.6<br />
O<br />
O<br />
293 K<br />
233 K<br />
218 K<br />
217 K<br />
216 K<br />
213 K<br />
Figure 7. 1 H-NMR spectra of 8 recorded at different temperatures (CDCl3, 300 MHz)<br />
3.4<br />
1.3. STEREOCHEMICAL ASPECTS OF BENZO-SPIRANES IN SOLID STATE<br />
The solid-state molecular structure was determined for 7 (Figure 8) using X-ray<br />
diffractometry on a monocrystal. The cyclohexane ring exhibits chair conformation having the<br />
O-<strong>Ph</strong> formal substituents in equatorial orientation, while the O-CH2 substituents are disposed in<br />
axial positions. The 1,3-dioxine rings have a twisted chair conformation, with the O2 and O14,<br />
respectively, outside the plane of the benzene (Figure 9). The initial assumption that the oxygen<br />
and spirane carbon atoms are in the same plane was close to reality, taking into account that the<br />
dihedral angle between O1-C10-O9 plane and O13-C14-O22 plane is 3.35°. The two benzene<br />
rings are oriented in anti conformation and are parallel (dihedral angle 1.471°).<br />
Figure 3. ORTEP 13 drawing of 7<br />
The package of molecules in the monocrystal caused an interesting stacking pattern by<br />
the interaction between one benzene ring and one hydrogen atom belonging to the methylene<br />
group of the 1,3-dioxine moiety of a neighboring molecule (Figure 4). The distance between this<br />
hydrogen and the centre of the neighboring phenylene is 2.66 Å.<br />
13 Farrugia, L.J. J Appl Cryst 1997, 30, 565.<br />
O<br />
3.2<br />
O<br />
3.0<br />
2.8<br />
2.6<br />
2.4<br />
2.2<br />
2.0<br />
1.8<br />
1.6<br />
8
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
Figure 4. Mercury drawing of two neighboring molecules in the monocrystal of 7<br />
2. SYN<strong>THESIS</strong>, <strong>STEREOCHEMISTRY</strong> <strong>AND</strong> ADSORPTION STUDIES OF SOME NEW<br />
(POLY)SPIRANES CONTAINING 1,2-DITHIOLANE UNITS<br />
2.1. The Synthesis<br />
The synthesis 14,15 of the target (poly)spiranes involves as the first step the (a)cetalization<br />
reaction of various carbonyl compounds with 2,2-bis(bromomethyl)-1,3-propanediol (Scheme 7).<br />
The equilibrium of the reaction was shifted toward products by removing the water by azeotropic<br />
distillation of the reaction water with the solvent (benzene or toluene).<br />
R<br />
R1<br />
HO<br />
Br benzene, reflux R1 O<br />
O +<br />
R2 HO<br />
Br APTS R2 O<br />
9a-d<br />
10<br />
1 - 8 hrs.<br />
11-15<br />
O +<br />
HO<br />
Br benzene, reflux<br />
R<br />
O<br />
HO<br />
Br pTSA<br />
O<br />
16a-c<br />
10<br />
3 - 10 hrs.<br />
17-19<br />
O O<br />
20<br />
HO<br />
HO<br />
Br<br />
Br<br />
CHO<br />
+<br />
HO<br />
Br benzene, reflux<br />
CHO HO<br />
Br pTSA<br />
3 - 10 hrs.<br />
O O<br />
22a, o-formyl<br />
22b, m-formyl<br />
+ 2<br />
10<br />
10<br />
benzene, reflux<br />
pTSA<br />
6 hrs.<br />
Br<br />
Br<br />
Br<br />
Br<br />
Scheme 7<br />
Br<br />
Br<br />
O<br />
O<br />
Br Br<br />
R1 R2 Carbonyl Compound Product<br />
CH 3 CH 3 9a 11<br />
H C 6 H 5 9b 12<br />
H 1-C 10 H 7 9c 13<br />
CH 3 C 6 H 5 9d 14<br />
CH 3 1-C 10 H 7 9e 15<br />
R Ketone Product<br />
H 16a 17<br />
C 6 H 5 16b 18<br />
CH 3 16c 19<br />
21<br />
O<br />
O<br />
O<br />
O<br />
23: ortho<br />
24: meta<br />
The assigned structures were confirmed by NMR spectra.<br />
For the obtainment of the target cyclic disulfides 25-32 we have modified a procedure<br />
previously reported by Chorbadijev et al 16 for the synthesis of dome acyclic disulfide derivatives<br />
(Scheme 8) via substitution of a halide atom with disodium disulfide (previously generated from<br />
14<br />
Gropeanu, R.A.; Woiczechowski-Pop, A.; Tintas, M.; Turdean, R.; Grosu, I. Studia Univ. Babes-Bolyai, Ser.<br />
Chim. 2005, 50, 247.<br />
15<br />
Gropeanu, R.A.; Tintas, M.; Pilon, C. ; Morin, M.; Breau, L.; Turdean, R.; Grosu, I. “SYN<strong>THESIS</strong>,<br />
<strong>STEREOCHEMISTRY</strong> <strong>AND</strong> ADSORPTION STUDIES OF NEW (POLY)SPIRANES CONTAINING 1,2-<br />
DITIOLANE UNITS “Monatshefte für Chemie 2005, submitted.<br />
16<br />
Chorbadjiev, S.; Roumian, C.; Markov, P. J. Prakt. Chem. 1977, 319, 1036<br />
Br<br />
Br<br />
Br<br />
Br<br />
9
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
sulphur and sodium hydroxide). In order to increase the yield of the cyclic disulfide via<br />
suppression of polymer generation we greatly reduced the reagent concentrations (from 0.5 M to<br />
0.03M).<br />
Br<br />
Br<br />
R 1<br />
R 2<br />
EtOH, reflux 6 NaOH + 6 S 2 Na2S2 + Na2S2O3 + 3 H2O<br />
1 hr.<br />
O<br />
O<br />
Br<br />
Br<br />
+ Na2S2<br />
11: R1 = CH3, R2 = CH3<br />
12: R 1 = H, R 2 = C 6H 5<br />
14: R 1 = CH 3, R 2 = C 6H 5<br />
15: R 1 = CH 3, R 2 = 1-C 10H 7<br />
EtOH, reflux R O<br />
4 h O<br />
1<br />
R 2<br />
9 10 1<br />
2<br />
S<br />
8<br />
5 S<br />
3<br />
7 6 4<br />
25: R1 = CH3, R2 = CH3<br />
26: R 1 = H, R 2 = C 6H 5<br />
27: R 1 = CH 3, R 2 = C 6H 5<br />
28: R1 = CH3, R2 = 1-C10H7<br />
R<br />
O<br />
O<br />
Br<br />
Br<br />
+ Na2S2 EtOH, reflux 4 h<br />
12<br />
R<br />
11<br />
13 14<br />
O<br />
8<br />
O<br />
15 1<br />
5<br />
2<br />
S<br />
S<br />
3<br />
10 9 7 6 4<br />
O<br />
17: R = H<br />
18: R = C 6H 5<br />
19: R = CH 3<br />
O<br />
O O<br />
Br<br />
Br<br />
+ Na2S2<br />
29: R = H<br />
30: R = C 6H 5<br />
31: R = CH 3<br />
EtOH, refl ux<br />
17<br />
S<br />
18 19 20 21<br />
O<br />
22 23<br />
O<br />
24 1<br />
2<br />
S<br />
4 h S<br />
16<br />
14<br />
15 13<br />
O11<br />
12 10<br />
8 O<br />
9 7<br />
5<br />
6 4<br />
S<br />
3<br />
21 32<br />
Scheme 8<br />
The target (poly)spiranes afforded after the flash chromatography in fair to good yields (35-74%<br />
for 25-31 and 27% for 32). The mass spectra and NMR spectra of the products confirmed the<br />
assigned structures.<br />
2.2. The Stereochemistry of (Poly)spiranes Containing 1,2-Dithiolane Units<br />
Monospirane 25, dispirane 29 and tetraspirane 32, that are showing symmetrical substituted sixmembered<br />
rings, exhibit flexible structures. The six-membered rings prefer the chair<br />
conformation. Taking into account the literature data concerning the conformation of 1,2dithiolane<br />
ring we propose for this heterocycle, in our compounds, the envelope conformer with<br />
the spirane atom out of the plane. The flipping of the 1,3-dioxane (A) and 1,2-dithiolane (B)<br />
rings in 25 (Scheme 9) renders equivalent in NMR positions 1 with 4 and 6 with 10. The 1 H-<br />
NMR spectrum at rt do not discriminate the axial and equatorial orientations of the protons or of<br />
the similar group and exhibits only three singlets (δ1,4 = 2.94, δ6,10 = 3.71, δ8-Me = 1.37 ppm).<br />
Compound 29 shows four isomers of configuration, due to the chirality of the spiranes units with<br />
six-membered rings 17,18 and to the syn or anti arrangement of the peripheral rings for di or<br />
polyspirane skeleta. 19,20,21 The flipping of 1,3-dioxane, cyclohexane and 1,2-dithiolane rings<br />
(Scheme 10) determines the conformational equilibrium of all the possible isomers. The 1 H-<br />
NMR spectrum of 29 presents only two singlets for the protons of 1,3-dioxane and 1,2-dithiolane<br />
rings (δ1,4 = 2.96, δ6,15 = 3.74 ppm). The flipping of the peripheral 1,2-dithiolane rings and of the<br />
middle six-membered cycles of tetraspirane 32 determines the equilibrium of all possible<br />
isomers. The 1 H-NMR spectrum of 32 is very simple and shows only three singlets (δ1,4,15,18 =<br />
3.00, δ6,13,19,24 = 3.76, δ9,10,21,22 = 1.87 ppm).<br />
17<br />
Grosu, I.; Mager, S.; Ple, G.; Horn, M. J. Chem. Soc., Chem. Commun. 1995, 167.<br />
18<br />
Grosu, I.; Mager, S.; Ple, G. J. Chem. Soc. Perkin. Trans. 2 1995, 1351.<br />
19<br />
Grosu, I.; Mager, S.; Ple, G.; Mesaros, E. Tetrahedron 1996, 52, 12783.<br />
20<br />
Opris, D.; Grosu, I.; Toupet, L.; Ple, G.; Terec, A.; Mager, S.; Muntean, L. J. Chem. Soc., Perkin Trans. 1 2001,<br />
2413.<br />
21<br />
Terec, A.; Grosu, I.; Condamine, E.; Breau, L.; Ple, G.; Ramondenc, Y.; Rochon, F.D.; Peulon-Agasse, V.; Opris,<br />
D. Tetrahedron 2004, 60, 3173.<br />
10
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
12<br />
11<br />
A<br />
A<br />
O<br />
O<br />
H3C<br />
9<br />
10<br />
O<br />
O A<br />
2<br />
1<br />
S<br />
3<br />
B S<br />
5 4<br />
8 7 6<br />
CH3<br />
H 3C<br />
B<br />
B<br />
syn (P)<br />
O<br />
O<br />
III<br />
C<br />
B<br />
anti (P)<br />
II<br />
CH 3<br />
O<br />
O<br />
C S<br />
S<br />
C S<br />
S<br />
A<br />
A<br />
B<br />
B S<br />
S<br />
A<br />
B<br />
B<br />
25<br />
Scheme 9<br />
O<br />
O<br />
B<br />
anti (M)<br />
IV<br />
H3C<br />
H 3C<br />
O<br />
O<br />
CH3<br />
CH 3<br />
S<br />
C S<br />
A<br />
A<br />
O A<br />
O<br />
A<br />
A<br />
S<br />
B S<br />
B S<br />
S<br />
1 2<br />
S<br />
S<br />
13<br />
A<br />
14<br />
15<br />
O<br />
O B<br />
8 7 6<br />
9 syn (M)<br />
3<br />
C S<br />
5 4 A<br />
A<br />
O<br />
O B<br />
anti (P)<br />
C S<br />
C<br />
A<br />
O<br />
O B<br />
syn (P)<br />
10 I II III<br />
29<br />
Scheme 10<br />
Spiranes 26-28, with differently substituted 1,3-dioxane units exhibit anancomeric 1,3dioxane<br />
rings. The flipping of the heterocycle is shifted towards the conformation in which the<br />
most bulky substituent is placed in equatorial position (Scheme 11; <strong>Ph</strong> for 26 and Me for 27 and<br />
28; for similar cases see references 22,23,24 ). The flipping of the 1,2-dithiolane ring renders<br />
equivalent in NMR positions 6 and 10. However the 1 H-NMR spectra of 26-27 are more<br />
complex and exhibit different signals for the equatorial and axial protons at positions 6 and 10<br />
and for the axial and equatorial methylene groups of the 1,2-dithiolane ring (e.g. compound 26:<br />
δ6a,10a = 3.90, δ6e10e = 4.13, δCH2a = 3.49, δCH2e = 2.71 ppm).<br />
R<br />
O<br />
O<br />
V VI<br />
2<br />
R 1<br />
S<br />
S<br />
R<br />
O<br />
O 2<br />
R 1<br />
A<br />
B<br />
A<br />
A<br />
S<br />
S<br />
O<br />
R O 2<br />
R 1<br />
O<br />
O<br />
R 1<br />
R 2<br />
2<br />
1<br />
9<br />
3<br />
10 B<br />
5 4 A<br />
8 7 6<br />
VI'<br />
B<br />
A<br />
VI"<br />
26: R 1 = H, R 2 = C6H5<br />
27: R 1 = C6H5, R 2 = CH3<br />
28: R 1 = 1-C10H7, R 2 = CH3<br />
Scheme 11<br />
O<br />
O<br />
S<br />
B S<br />
O<br />
B<br />
syn (P)<br />
O<br />
A<br />
B<br />
anti (M)<br />
22 Anteunis, M.J.O.; Tavernier, D.; Borremans, F. Heterocycles 1976, 4, 293.<br />
23 Mager, S.; Grosu, I. Stud. Univ. “Babes-Bolyai”, Ser. Chemia 1988, 33, 47.<br />
24 Grosu, I.; Plé, G.; Mager, S.; Mesaros, E.; Dulau, A.; Gego, C. Tetrahedron 1998, 54, 2905.<br />
S<br />
B S<br />
III<br />
IV<br />
S<br />
C S<br />
C S<br />
S<br />
C S<br />
S<br />
11
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
Dispiranes 30 and 31 display semiflexible structures (Scheme 12), the cyclohexane ring<br />
is rigid, the substituent of this cycle being an efficient “holding group” and the 1,3-dioxane and<br />
1,2-dithiolane parts are flipping and render in equilibrium the possible stereoisomers (VII-X).<br />
The 1 H-NMR spectra do not differentiate the axial or equatorial orientation of the protons of the<br />
heterocycles but exhibit different signals (singlets) for the diastereotopic positions 6 and 15 (30:<br />
δ6 = 3.81, δ15 = 3.85, δ1,4 = 3.04 ppm and 31: δ6 = 3.71, δ15 = 3.76, δ1,4 = 2.98 ppm). The<br />
diastereotopicity of positions 6 and 15 is observed in 13 C-NMR spectra, too (30: δ6 = 66.32, δ15 =<br />
66.50 ppm and 31: δ6 = 66.34, δ15 = 66.61 ppm). Position 6 is procis and position 15 is protrans<br />
referred to the substituent at position 11.<br />
12<br />
11<br />
R 1<br />
R 1<br />
13<br />
A<br />
10<br />
A<br />
1 2<br />
S<br />
14<br />
15<br />
O<br />
O B<br />
3<br />
C S<br />
5 4<br />
8 7 6<br />
9<br />
syn (M)<br />
VII<br />
O<br />
O<br />
C<br />
B<br />
S<br />
C S<br />
B<br />
B<br />
R 1<br />
R 1<br />
A<br />
O B<br />
O<br />
syn (P)<br />
anti (M) anti (P)<br />
30, 31<br />
VIII<br />
X<br />
Scheme 12<br />
Figure 5. 1 H-NMR spectrum of 30 (CDCl3, 300 MHz, r.t.)<br />
2.3. ADSORPTION STUDIES<br />
A. Adsorption on planar gold surfaces<br />
Individual gold (111) surfaces were immersed in 1 mM ethanolic solutions of the cyclic<br />
disulfides, 29 to 32, for at least 24 hours in order to form monolayers. The monolayers were then<br />
analyzed by reflective IR spectroscopy. The relevant fragments of the spectra of two compounds<br />
are depicted in Figures 6 and 7. The wave numbers of the vibrational bands of their IR spectra<br />
are presented in Table 2. The assignment of the vibrational bands and the direction and<br />
magnitudes of the vibrational dipole moments were obtained from Hartree-Fock calculations<br />
done with Gaussian 98® software.<br />
O<br />
O<br />
H6, H15<br />
A<br />
15<br />
1<br />
6<br />
4<br />
S<br />
S<br />
O<br />
IX<br />
O<br />
C<br />
B<br />
C S<br />
S<br />
C S<br />
S<br />
H1, H4<br />
12
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
Absorbance<br />
0.0008<br />
0.0006<br />
0.0004<br />
0.0002<br />
0.0000<br />
-0.0002<br />
( ) ( q )<br />
2700 2800 2900 3000 3100 3200<br />
Wavenumbers (cm -1 )<br />
1<br />
O<br />
O<br />
S<br />
S<br />
Absorbance<br />
0.001<br />
0.000<br />
-0.001<br />
900 1000 1100 1200 1300 1400 1500 1600 1700 1800<br />
Wavenumbers (cm -1 )<br />
Figure 6. IR reflexion-absorption spectrum of a monolayer of 31 adsorbed on a gold (111) surface (relevant<br />
fragments)<br />
Absorbance<br />
0.0002<br />
0.0000<br />
3<br />
S<br />
O O<br />
S O O<br />
2700 2800 2900 3000 3100 3200<br />
Wavenumbers (cm -1 )<br />
S<br />
S<br />
Absorbance<br />
0.0005<br />
0.0000<br />
900 1000 1100 1200 1300 1400 1500 1600 1700 1800<br />
Wavenumbers (cm -1 )<br />
1<br />
O<br />
O<br />
3<br />
S<br />
O O<br />
S O O<br />
Figure 7. IR reflexion-absorption spectrum of a monolayer of 32 adsorbed on a gold (111) surface (relevant<br />
fragments)<br />
The widths of the bands at half-height in the IR spectra are in the range of 20-40 cm -1<br />
which suggests that the molecules are disordered on the gold surface. Also, the methylene<br />
νa(CH2) band at 2926-2930 cm -1 (that is present in all samples) is indicative of a liquid like<br />
monolayer. This disorder is probably due to the complex and relatively rigid structure of the<br />
molecules which results in a large distance between the adsorbed molecules and thus few<br />
stabilizing inter-adsorbate interactions that usually results in self-assembly. Although the<br />
monolayer is not very organized, some information on the orientation of the adsorbed molecules<br />
could be drawn from the IR investigations. Our analysis of the IR spectra are based on the<br />
direction of the vibrational dipole moments obtained from a computational chemistry calculation<br />
and the infrared surface selection rule for adsorbates on metallic substrates 25 which states that a<br />
vibrational mode is IR active if there is a component of its dipole moment perpendicular to the<br />
surface.<br />
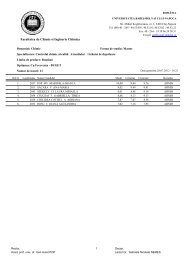
Table 2. IR wave numbers (ν/cm -1 ) of the SAMs for each cyclic disulfide 29-32 on a gold (111)<br />
surface<br />
Assignment 29 30 31<br />
32<br />
νa(=C-H, Ar) - 3065 (weak) - -<br />
νa(=C-H, Ar) - 3031 (weak) - -<br />
νa(=C-H, Ar)<br />
νa(CH3)<br />
-<br />
2961<br />
3017 (weak) - -<br />
a (weak) 2959 a<br />
2958 2958 a (weak)<br />
νa(CH2)<br />
νs(CH3)<br />
2928<br />
-<br />
2930<br />
2874<br />
2926 2926<br />
a<br />
2869 -<br />
νs(CH2) 2857 2856 2857 2857<br />
νa(C-O-C) 1096 1131 1154 1130<br />
a<br />
due to a small amount contaminant containing a methyl group<br />
The analysis of the wave numbers and intensity of the vibrational bands of compounds 30<br />
and 32 adsorbed on gold suggest that they adopt the orientations shown in Figures 19 and 20.<br />
25 Greenler, R.G. J. Chem. <strong>Ph</strong>ys. 1966, 44, 310.<br />
S<br />
S<br />
S<br />
S<br />
13
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
Also, it is likely that both compounds bind to the surface via at least two of their sulphurs, as<br />
disulfides are known to dissociate upon adsorption on gold. 26<br />
The absence of CH deformation bands between 1300 and 1400 cm -1 suggests that 32 is<br />
not oriented perpendicular to the surface but is rather parallel to the surface. In this orientation<br />
the C-H deformation modes of methylenes will have a low intensity. It is thus possible that<br />
compound 32 is anchored to the surface by both 1,2-dithiolane rings. However, this would<br />
require an important reorganization of the optimized structure shown in Figure 8.<br />
a b c<br />
Figura 8. Possible orientations of compounds 30 (a) and 32 (b) and (c) adsorbed on Au(111) suggested by the<br />
analysis of its IR spectra.<br />
B. Adsorption on gold electrodes and interaction with gold nanoparticles<br />
The interaction between some (poly)spiranes having 1,2-dithiolane units and gold<br />
nanoparticles (GNP) was studied for the beginning using UV-Vis spectroscopy. Thus, the UV-<br />
Vis spectra of the organic ligands in chloroform and of the GNP (water solution) were recorded,<br />
as well as the spectra of the samples obtained by mixing cyclic disulfides with GNP solution<br />
(Figure 9). The main feature of the samples spectra is the decay of the plasmodic resonance peak.<br />
For example, in short time (20 minutes) the absorbance in the 500-550 nm region of the spectra<br />
was five times reduced in the case of interaction between 32 and GNP (Figure 9d). Also, the<br />
electronic absorption peak of the organic ligand (330 mn) has disappeared (Figure 9c).<br />
Absorbanta<br />
0.12<br />
0.11<br />
0.10<br />
0.09<br />
0.08<br />
0.07<br />
0.06<br />
0.05<br />
0.04<br />
440 460 480 500 520 540 560 580 600 620<br />
λ(nm)<br />
Absorbanta<br />
4.0<br />
3.5<br />
3.0<br />
2.5<br />
2.0<br />
1.5<br />
1.0<br />
0.5<br />
0.0<br />
-0.5<br />
100 200 300 400 500 600 700 800 900 1000<br />
λ(nm)<br />
Absorbanta<br />
4.0<br />
3.5<br />
3.0<br />
2.5<br />
2.0<br />
1.5<br />
1.0<br />
0.5<br />
0.0<br />
-0.5<br />
200 300 400 500 600<br />
λ(nm)<br />
Absorbanta<br />
0.042<br />
0.040<br />
0.038<br />
0.036<br />
0.034<br />
0.032<br />
0.030<br />
0.028<br />
0.026<br />
0.024<br />
0.022<br />
Au+STS-1min<br />
Au+STS-5min<br />
Au+STS-20 min<br />
0.020<br />
440 460 480 500 520 540 560 580 600 620<br />
a b c d<br />
Figure 9. UV-Vis spectra of the a) water dispersion of GNPs, b) chloroform solution of 32, c) GNPs<br />
functionalized with 32, and d) GNPs functionalized with 32, recorded at three different times<br />
These facts suggest an aggregation process of the nanoparticles, which is sustained by the<br />
transmission electronic microscope (80 kV) measurements (Figures 10).<br />
a b c<br />
Figure 10. Transmission electronic microscope image of the a) GNPs suspension, b) GNPs functionalized with<br />
30, and c) GNPs functionalized with 32<br />
26 Fenter, P.; Eberhardt, A.; Eisenberg, P. Science 1994, 266, 1216<br />
λ(nm)<br />
14
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
A method which is suitable for the study of the adsorption of the cyclic disulfides on the<br />
gold surface is the cyclic voltammetry using a gold electrode as working electrode. After the<br />
cleaning operations, the cyclic voltammetry of the standard system [K4Fe(CN)6 in 0.1 M KCl,<br />
Figure 11a] was performed and compared with the cyclic voltammograms run with the gold<br />
electrodes functionalized with each of cyclic disulfide 25-27, 30, 32. The absence of a redox<br />
process has shown that the spirane monolayer adsorbed on the surface of the gold electrode acts<br />
as a dielectric isolator (Figure 11b).<br />
a b<br />
Figure 11. Cyclic voltammogram of a solution of 3 mM K4Fe(CN)6 in 0.1 M KCl using a) bare gold electrode<br />
(10 mV/sec) and b) urface functionalized gold electrode with 32 (50 mV/sec)<br />
31 has two cyclic disulfide units per molecule and, therefore, was tested for the<br />
functionalization of the gold electrode surface with GNP. Two methods of the preparation were<br />
tried: Method A: the electrode treated with the organic ligand was immersed in the GNP solution,<br />
and Method B: the electrode was immersed into the solution obtained by mixing of GNP with<br />
organic ligand at different pH values (5.5, 7 and 11).<br />
I / mA<br />
I / mA<br />
0.04<br />
0.03<br />
0.02<br />
0.01<br />
0.00<br />
-0.01<br />
-0.02<br />
-0.03<br />
0.06<br />
0.04<br />
0.02<br />
0.00<br />
-0.02<br />
-0.04<br />
0.0 0.1 0.2 0.3 0.4 0.5 0.6<br />
Potential /V vs. Ag/AgCl<br />
0.0 0.1 0.2 0.3 0.4 0.5 0.6<br />
Potential / V vs. Ag/AgCl<br />
-0.6 -0.4 -0.2 0.0 0.2 0.4 0.6<br />
a b<br />
Figure 12. Cyclic voltammograms of a) a solution of 3 mM K4Fe(CN)6 in 0.1 M KCl (buffered at pH<br />
5.5) using surface functionalized gold electrode with 32 and GNP (10 minutes immersion, pH 7, by<br />
Method A (50 mV/sec)), and b) a solution of 3 mM K4Fe(CN)6 in 0.1 M KCl (buffered at pH 7) using<br />
surface functionalized gold electrode with 32 and GNP (pH 7) by Method B (50 mV/sec)<br />
These functionalized electrodes were subjected to cyclic voltammetry of the standard<br />
solution. The results showed us that only the Method B run at pH 7 was successful (Figure 12),<br />
the voltammograms presenting a reversible redox process. Comparing the voltammograms, we<br />
can remark that for the GNP-32-modified gold electrode the current maxima are more than two<br />
times higher that the corresponding peaks for the non-functionalized gold electrode, meaning<br />
that the modification of the surface is increasing the electron transfer between solution and<br />
electrode.<br />
I / mA<br />
0.025<br />
0.020<br />
0.015<br />
0.010<br />
0.005<br />
0.000<br />
-0.005<br />
-0.010<br />
I / mA<br />
0.0 0.1 0.2 0.3 0.4 0.5 0.6<br />
0.15<br />
0.10<br />
0.05<br />
0.00<br />
-0.05<br />
-0.10<br />
Potential / V vs. Ag/AgCl<br />
Potential / V, vs. Ag/AgCl<br />
15
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
PART B.<br />
SYN<strong>THESIS</strong> <strong>AND</strong> PHYSICO-CHEMICAL PROPERTIES OF SOME NEW<br />
MACROCYCLES CONTAINING 10H-PHENOTHIAZINE UNITS<br />
1. INTRODUCTION<br />
<strong>Ph</strong>enothiazines belong to a pharmaceutically important class of heterocycles, 27 and due to<br />
their pharmacological efficacy they are applied in a broad range as sedatives, tranquilizers,<br />
antituberculotics, antipyretics, antitumor agents, bactericides and parasiticides. 28 They are also<br />
able to cleave DNA upon photochemical induction. 29 As a consequence of a low oxidation<br />
potential, these tricyclic nitrogen-sulfur heterocycles readily form stable radical cations and a<br />
key role of their physiological activities can be attributed to this circumstance. 30 An interesting<br />
feature of phenothiazines is the fact that the first oxidation step is reversible. 31 Thus,<br />
phenothiazine derivatives have become spectroscopic probes in molecular and supramolecular<br />
arrangements for photoinduced electron transfer (PET) studies. 32<br />
Our work in this field had the following goals:<br />
- the synthesis of some new building blocks useful in the preparation of oligophenothiazines of<br />
determined number of phenothiazine units<br />
- the synthesis of phenothiazine-ferrocene dyads and triads and to study the electronic<br />
communication between the redox-active units of the oligomers, and of ferrocene-phenothiazine<br />
cyclophane<br />
- the synthesis of phenothiazine-containing macrocycles (shape-persisted macrocycles, as well as<br />
phenothiazine coronands).<br />
2. ORIGINAL RESULTS<br />
2.1. SYN<strong>THESIS</strong> OF IODO-PHENOTHIAZINE DERIVATIVES<br />
The corner stone of our strategies was 10-alkyl-3,7-dibromo-10H-phenothiazine (1),<br />
which is readily available from commercially 10H-phenothiazine (1) in two steps: the first step is<br />
represented by an alkylation in the position 10 (at the secondary nitrogen of the phenothiazine<br />
27<br />
Sainsbury, M. In Comprehensive Heterocyclic Chemistry, Vol. 3; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon:<br />
Oxford, 1984, 995<br />
28<br />
(a) Mietzsch, F. Angew. Chem. 1954, 66, 363. (b) Ionescu, M.; Mantsch, H. Adv. Heterocycl. Chem. 1967, 8, 83.<br />
(c) Bodea, C.; Silberg, I. Adv. Heterocycl. Chem. 1968, 9, 321. (d) Valzelli, L.; Garattini, S. In Principles of<br />
Psychopharmacology; Clark, W. G., Ed.; Academic Press: 1970, 255. (e) Okafor, C. O. Heterocycles 1977, 7, 391.<br />
(f) Eckstein, Z.; Urbanski, T. Adv. Heterocycl. Chem. 1978, 23, 1. (g) Szabo, J. Chem. Heterocycl. Compd. USSR<br />
(Engl. Trans.) 1979, 15, 291. (h) Albery, W. J.; Foulds, A. W.; Hall, K. J.; Hillman, A. R.; Edgell, R. G.; Orchard,<br />
A. F. Nature 1979, 282, 793.<br />
29<br />
(a) Nishiwaki, E.; Nakagawa, H.; Takasaki, M.; Matsumoto, T.; Sakurai, H.; Shibuya, M. Heterocycles 1990, 31,<br />
1763.<br />
(b) Decuyper, J.; Piette, J.; Lopez, M.; Merville, M. P.; Vorst, A. Biochem. <strong>Ph</strong>armacol. 1984, 33, 4025. (c) Motten,<br />
A. G.; Buettner, G. R.; Chignell, C. F. <strong>Ph</strong>otochem. <strong>Ph</strong>otobiol. 1985, 42, 9. (d) Fujita, H.; Matsuo, I. Chem. Biol.<br />
Interac. 1988, 66, 27.<br />
30<br />
(a) Forrest, I.; Forrest, F. Biochim. Biophys. Acta 1958, 29, 441. (b) Iida, Y. Bull. Chem. Soc. Jpn. 1971, 44, 663.<br />
(c) Moutet, J.-C.; Reverdy, G. Nouv. J. Chim. 1983, 7, 105.<br />
31<br />
For cyclovoltammetric and spectroscopic data of phenothiazine, see for example: Tinker, L. A.; Bard, A. J. J. Am.<br />
Chem. Soc. 1979, 101, 2316. (d) Padusek, B.; Kalinowski, M. K. Electrochim. Acta 1983, 28, 639. (e) McIntyre, R.;<br />
Gerischer, H. Ber. Bunsen Ges. <strong>Ph</strong>ys. Chem. 1984, 88, 963.<br />
32<br />
(a) Duesing, R.; Tapolsky, G.; Meyer, T. J. J. Am. Chem. Soc. 1990, 112, 5378. (b) Jones, W. E. Jr.; Chen, P.;<br />
Meyer, T. J. J. Am. Chem. Soc. 1992, 114, 387. (c) Brun, A. M.; Harriman, A.; Heitz, V.; Sauvage, J.-P. J. Am.<br />
Chem. Soc. 1991, 113, 8657. (d) Burrows, H. D.; Kemp, T. J.; Welburn, M. J. J. Chem. Soc., Perkin Trans. 2 1973,<br />
969. (e) Collin, J.-P.; Guillerez, S.; Sauvage, J.-P. J. Chem. Soc., Chem. Commun. 1989, 776. (f) Daub, J.; Engl, R.;<br />
Kurzawa, J.; Miller, S. E.; Schneider, S.; Stockmann, A.; Wasielewski, M. R. J. <strong>Ph</strong>ys. Chem. A 2001, 105, 5655.<br />
16
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
nucleus, Scheme 1). After the purification, the intermediate 3 is subjected to a bromination 33 to<br />
afford the dibromo-phenothiazine 1 in excellent yield (98-99%, 69-79% overall yield).<br />
8<br />
7<br />
H<br />
9 10<br />
N<br />
6<br />
S<br />
5<br />
2<br />
1<br />
4<br />
2<br />
3<br />
1° 1.1 equiv. t-BuOK/1,4-dioxane<br />
10 min. r.t., then 10 min. reflux<br />
2° 1.1equiv. Alkyl-Br,<br />
overnight, r.t.<br />
60-80%<br />
Alkyl<br />
N<br />
S<br />
3<br />
Scheme 1<br />
2.1 equiv. Br 2 /AcOH<br />
overnight, r.t.<br />
98-99%<br />
Alkyl<br />
N<br />
Br S<br />
Br<br />
1a, Alkyl = C 2 H 5<br />
1b, Alkyl = n-C 6 H 13<br />
The synthesis of the iodine-phenothiazines was realized 34 by a mono or a double Li-Br<br />
exchange with nBuLi (Scheme 2) and a subsequent addition of iodine as electrophile.<br />
Alkyl<br />
N<br />
2° 2.1 equiv. I<br />
Br S<br />
Br 2<br />
I S<br />
I<br />
1a, Alkyl = C 2 H 5<br />
1b, Alkyl = n-C 6 H 13<br />
1° 2.1 equiv. nBuLi/THF/-78°C, 30 min.<br />
Scheme 2<br />
Alkyl<br />
N<br />
4a, Alkyl = C 2 H 5<br />
4b, Alkyl = n-C 6 H 13<br />
The iodine-phenothiazine structures of 4a, 4b were revealed by MS (each of the FAB-MS<br />
spectra present the molecular peak, i.e. for 4b M = 535, and M/z = 534.9) and by NMR<br />
measurements. The 1 H-NMR spectra have shown that the products are symmetrical substituted<br />
phenothiazine derivatives, because of the occurrence in the aromatic region of three distinct<br />
signals corresponding to the three pairs of phenothiazine aromatic protons.<br />
One interesting feature of the iodo-derivatives is the upfield shifting of the signal of the<br />
direct bounded carbon atom, compared to the bromine-substituted one. This shift is called<br />
“heavy-atom effect”. For our compounds, the assigned chemical shift for this type of carbon<br />
atom is 84.8 ppm (Figure 1).<br />
200<br />
175<br />
8<br />
9<br />
150<br />
a<br />
N<br />
I S<br />
I<br />
6<br />
4<br />
1<br />
2<br />
125<br />
100<br />
C3, C7<br />
Figure 1. 13 C-NMR spectrum of 4b (CD3COCD3, 300 MHz, r.t.)<br />
33 Bodea, C; Terdic, M. Studii si Cercet. Chimie, Acad. RPR Fil. Cluj 1962, 13, 81-87.<br />
34 Sailer, M.; Gropeanu, R.A.;. Müller, T.J.J. “PRACTICAL SYN<strong>THESIS</strong> OF IODO PHENOTHIAZINES. A<br />
FACILE ACCESS TO ELECTROPHORE BUILDING BLOCKS” Journal Of Organic Chemistry 2003, 68, 7509.<br />
75<br />
50<br />
25<br />
0<br />
17
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
The success of this reaction prompted us to investigate the possibility of the introduction<br />
of one iodine atom in the phenothiazine molecule. This was realized by performing of a mono<br />
Br-Li exchange by addition of only one equivalent of nBuLi in much longer time over of a more<br />
diluted solution of 1 (Scheme 3).<br />
Alkyl<br />
N<br />
2° 1 equiv. I<br />
Br S<br />
Br 2<br />
I S Br<br />
1b, Alkyl = n-C 6 H 13<br />
1° 1 equiv. nBuLi/THF/-78°C, 30 min.<br />
Scheme 3<br />
Alkyl<br />
N<br />
5, Alkyl = n-C 6 H 13<br />
The FAB-MS spectrum of 5 presents the molecular peak and its characteristic splitting of<br />
the mono-brominated compounds (M/z = 488.7, 486.7, M=488). The NMR spectra show an<br />
unsymmetrical substitution of the phenothiazine. Moreover, the 13 C-NMR spectrum presents a<br />
signal due to a quaternary carbon atom at 84.8 ppm, assigned to the iodine-substituted carbon<br />
atom.<br />
The above described method generates bromo-iodo-phenothiazines useful in the<br />
construction of oligophenothiazines. In order to obtain other unsymmetrical substituted iodophenothiazines<br />
that contain other functional groups it was necessary to develop a different<br />
method. Thus, we have relied on the Li-Br exchange and modified the electrophiles. Using 2<br />
equivalents of nBuLi and two kind of electrophiles (in principle, the first one iodine, and as the<br />
second one, other kind of electrophiles that can generate useful functions) could be obtained the<br />
target phenothiazine derivatives. This idea was tested for the synthesis of 3-iodo-10-hexyl-10Hphenothiazine<br />
(6, Scheme 4).<br />
N<br />
Br S<br />
Br<br />
1b<br />
1º 1 equiv. nBuLi/THF/-78ºC<br />
2º 1 equiv. H 2 O/THF/-78ºC<br />
80%<br />
Br<br />
1º 2 equiv. BuLi/THF/-78ºC<br />
2º 1 equiv. I 2 /THF/-78ºC<br />
3º 1 equiv. H 2 O/THF/-78ºC<br />
70%<br />
N<br />
S<br />
7<br />
Scheme 4<br />
I<br />
N<br />
S<br />
6<br />
1º 1 equiv. BuLi/THF/-78ºC<br />
2º 1 eqiiv. I 2 /THF/-78ºC<br />
The direct transformation of 4b into 6 was performed by using one equivalent of iodine (as the<br />
first added electrophile) and then water for quenching the dilithiated species of 4b. The<br />
sequential transformation of 4b involved the exchange of one bromine atom with hydrogen, via<br />
Li-Br exchange and the use of water as electrophile, and a subsequent replacement of the<br />
remaining bromine from 7 with iodine, using the same Li-Br exchange. Comparing the yields,<br />
these two pathways are quite similar, but the direct method brings some advantages: it reduces<br />
the materials consumption, as well as the working time.<br />
Having these on our mind, other electrophiles were also used in this procedure. Thus,<br />
D2O, dry DMF, B(OMe)3 were added as second electrophiles to afford the corresponding nonsymmetric<br />
substituted phenothiazines (Scheme 5).<br />
N<br />
88%<br />
Br S<br />
Br I S<br />
E<br />
4b 8-10<br />
E<br />
E+<br />
1º 2 equiv. BuLi/THF anh./-78ºC<br />
2º 1 equiv. I2/THF anh./-78ºC<br />
3º 1 equiv. E+/THF/-78ºC la t.c.<br />
8 9 10<br />
D CHO<br />
O<br />
B<br />
O<br />
D2O DMF B(OMe)3 + pinacole<br />
Scheme 5<br />
N<br />
18
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
The unsymmetrical structure of the phenothiazine derivatives 8-10 was shown by the NMR<br />
spectra. For example, the 1 H-NMR spectrum of 9 presents distinct 6 signals for the phenothiazine<br />
protons, as well as the signal corresponding for the formyl proton (δ 3-CHO = 9.80 ppm, Figure<br />
2).<br />
10.0<br />
9.5<br />
9.0<br />
8.5<br />
8.0<br />
7.5<br />
7.0<br />
6.5<br />
I S<br />
CHO<br />
Figure 2. 1 H-NMR spectrum of 9 (CD3COCD3, 300 MHz, r.t.)<br />
6.0<br />
N<br />
5.5<br />
The 13 C-NMR spectrum of 9 presents two characteristic signals: one corresponding to C-I (δ C7<br />
= 85.81 ppm) and one for the formyl group carbon atom (δ C3’ = 190.28 ppm).<br />
2.2. PALLADIUM-CATALYZED CROSS-COUPLING REACTIONS OF HALO-<br />
PHENOTHIAZINES<br />
Halo-phenothiazines, as many other halo-derivatives, 35 undergo palladium-catalyzed<br />
cross-coupling reactions. An extensively used method for the coupling of two Csp2 carbons is<br />
represented by the Suzuki cross-coupling. 36 This reaction involves the interaction of one organic<br />
halide and one organoboron compound, interaction mediated by Pd reduced species. This crosscoupling<br />
reaction undergo in the presence of charged bases (Na2CO3, K2CO3, Na3PO4, KOH, and<br />
alkoxides). 37,38<br />
The synthesis of the phenothiazine boronic intermediates was previously described. 39<br />
After generation of a mono or a double lithiated species by Li-Br exchange, an addition of<br />
trimethylborate as electrophile, followed by the transesterification with pinacole, afforded the<br />
phenothiazine mono- or bis-boronic esters (11a,b, 12, 13, Scheme 6). It was chosen the pinacole<br />
ester due to their high stability. 40<br />
In order to increase the selectivity, the reaction temperature was decreased. In the<br />
synthesis of 15, despite the very good selectivity, the yield was not very good, after work-up<br />
35 Poli, G.; Giambastiani, G.; Heumann, A. Tetrahedron 2000, 56, 5959.<br />
36 Miyaura, N.; Suzuki, A. Chem. Rev.1995, 95, 2457.<br />
37 Susuki, A. Acc. Chem. Res. 1982, 15, 178.<br />
38 Miyaura, N.; Yamada, K.; Suginome, H.; Suzuki, A. J. Am. Chem. Soc. 1985, 107, 972.<br />
39 Krämer, C.; Sailer, M.; Müller, T.J.J. Synthesis 2002, 1163.<br />
40 Hoffmann, R.W.; Dresely, S. Synthesis 1988, 103.<br />
5.0<br />
4.5<br />
4.0<br />
3.5<br />
3.0<br />
2.5<br />
2.0<br />
1.5<br />
1.0<br />
0.5<br />
19
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
some amounts of the starting materials were recovered. This was a sign that the temperature was<br />
too low. However, the synthesis of 14 has afforded in good yield the desired triad.<br />
N<br />
1° 2 equiv. nBuLi/THF/-78°C<br />
2° 2 equiv. B(OMe) 3 /THF/-78°C la t.c.<br />
3° 2 equiv. pinacol<br />
N<br />
Br S<br />
Br<br />
65%<br />
O<br />
B S<br />
B<br />
O<br />
4b O 11 O<br />
Br<br />
4b<br />
N<br />
S<br />
7<br />
1° 1 equiv. nBuLi/THF/-78°C<br />
2° 1 equiv. B(OMe) 3 /THF/-78°C la t.c.<br />
3° 1 equiv. pinacol<br />
75%<br />
1° 1 equiv. nBuLi/THF/-78°C<br />
2° 1 equiv. B(OMe) 3 /THF/-78°C la t.c.<br />
3° 1 equiv. pinacol<br />
80%<br />
Scheme 6<br />
N<br />
Br S<br />
B<br />
O<br />
12 O<br />
N<br />
O<br />
Br S<br />
B<br />
After the purification, only traces of the iodine-derivatives were isolated. The mass<br />
spectra (FAB-MS) of 14 and 15 confirmed the assigned structures. The molecular peaks are<br />
splitted; for example, for 14 (M = 1003) an usual distribution of M-2, M-1, M, M+1, M+2 peaks<br />
characteristic to the dibrominated compounds occurred in the spectrum.<br />
The 13 C-NMR spectra present characteristic peaks to the Csp2-Br at 114 ppm and no<br />
signals corresponding to a Csp2-I bond.<br />
Another dyad was synthesized in a different manner (Scheme 10), by an oxidative<br />
coupling of two monolithiated phenothiazines.<br />
N<br />
2<br />
Br S<br />
Br<br />
1b<br />
1° 1 equiv. BuLi/-78°C/THF<br />
2° dry CuCl 2<br />
Br<br />
Scheme 7<br />
2.3. FERROCENO-PHENOTHIAZINE MACROCYCLES<br />
Ferrocene represents an interesting redox-active material and, recently, the ferrocene core<br />
has been used as a building-block in the construction of redox-active and responsive<br />
macrocycles. 41<br />
Thus, the prospect of integrating strongly coupled redox fragments like phenothiazines<br />
and ferrocenes into conjugated chains could constitute a so far unknown class of redox<br />
addressable molecular wires. In particular, the integration of these synthons into a fullyconjugated<br />
macrocycle represented one of our main goals.<br />
Palladium-catalyzed cross-coupling reaction were described to be an effective method in<br />
the synthesis of ring-substituted ferrocenes. 42 From these methods, the Suzuki and the Negishi<br />
41 a) Dinnebier, R.E.; Ding, L.; Ma, K.; Neumann, M.A.; Tanpipat, N.; Leusen, F.J.J.; Stephens, P.W.; Wagner, M.<br />
Organometallics 2001, 20 (26), 5642, 5647; b) Beer, P.D.; Crowe, D.B.; Ogden, M.I.; Drew, M.G.B.; Main, B. J.<br />
Chem. Soc., Dalton Trans.: Inorg. Chem. 1993, (14), 2107-2116; c)Beer, P.D.; Nation, J.E.; Brown, S.L. J.<br />
Organomet. Chem. 1989, 377 (1), C23-C26; d) Bell, A.P.; Hall, D. J. Chem. Soc., Chem. Commun. 1980, (4), 163-<br />
165; e) Plenio, H.; Aberle, C. Chem. Eur. J. 2001, 7(20), 4438-4446; f) Plenio, E.; Aberle, C.; Al Shihadeh, Y.;<br />
Lloris, J.M.; Martinez-Manez, J.; Pardo, T.; Soto, J. Chem. Eur. J. 2001, 7(13), 2848-2861.<br />
S<br />
N<br />
13<br />
16<br />
N<br />
S<br />
O<br />
Br<br />
20
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
couplings provided good results and, therefore, these were taken into consideration for our<br />
purposes. Both of these methods involve the preparation of the dilithiated species of ferrocene 43<br />
(17, Scheme 8).<br />
Fe<br />
2.5 equiv. nBuLi<br />
1.25 equiv. TMEDA<br />
Fe<br />
Li<br />
1° 2 equiv. B(OMe) 3<br />
Fe<br />
Li<br />
2° pinacole or HO- /H2O 17<br />
2 equiv ZnCl 2<br />
Fe<br />
19<br />
ZnCl<br />
ZnCl<br />
Ar-X<br />
Pd(0) catalyst<br />
O<br />
B<br />
O<br />
O<br />
B<br />
O<br />
18a<br />
Fe<br />
Ar<br />
Ar<br />
where Ar = phenothiazine derivatives<br />
Scheme 8<br />
or<br />
Fe<br />
Ar-X<br />
Pd(0) catalyst<br />
The Suzuki cross coupling between 18a and 5 was performed using as catalyst<br />
tetrakis(triphenylphosphine)palladium and typical conditions; the conversion of the ferrocene<br />
18a was 100% (according to TLC), but the yield in the desired product was very low (about 1%).<br />
The “product” proved to be a mixture of at least two ferrocene derivatives (according to NMR<br />
and MS spectra). Using the MS (FAB+) spectrum we could assign the productsas mono-crosscoupling<br />
products (with bromo-substituted phenothiazine as the major product and iodosubtituted<br />
phenothiazine core as minor one) and the ferrocene mono-boronic-pinacole ester.<br />
The first attempts of Negishi coupling have furnished the desired products in fair to good<br />
yields. A series of Negishi couplings were performed in order to optimize the synthesis. The data<br />
are presented in Scheme 9 and in Table 1.<br />
The fact that the yields are rather modest could have two reasons:<br />
a) the bis(lithiated) ferrocene was described to be not very stable in the presence of<br />
ethers, such as THF or diethyleter. This means that is possible that the recovered ferrocene<br />
appeared as a decomposition process of the 1,1’-dilithium-ferrocene.<br />
b) the palladium catalyzed cross-coupling reactions between two rich electron systems<br />
proceeds usually with low yields.<br />
c) after all of the cross-coupling reactions there was recovered an insoluble, amorphous<br />
brown material, which is most probably a mixture of polymers.<br />
Analyzing the data, two conclusions could be drawn: the iodine-substituted<br />
phenothiazines react better than the brominated ones, that is not surprising. Unfortunately, this<br />
higher reactivity provides a larger amount of oligomeric by-products. Also, in the case of 4b<br />
bis(ferrocenyl)-phenothiazine (26) was recovered as by-product, and this was not the case for<br />
bromine-substituted phenothiazines. The second conclusion regards the catalysts: the best results<br />
were obtained using Pd[(P<strong>Ph</strong>3)4] for bromine-substituted phenothiazines, and PdCl2(P<strong>Ph</strong>3)2 for<br />
iodine-substituted phenothiazines, respectively.<br />
A Negishi cross coupling at higher temperature (reflux) was also performed, but the yield<br />
was lower than the reaction run at room temperature (see entry 9, Table 1).<br />
42 a) Arnold, R.; Matchett, S.A.; Rosenblum, M. Organometallics 1988, 7, 2261; b) Enders, M.; Kohl, G.; Pritzkow,<br />
H. J. Organomet. Chem. 2001, 622, 66-73; c) Enders, M.; Kohl, G.; Pritzkow, H. Organometallics 2002, 21, 1111-<br />
1117.<br />
43 a) Bishop, J.J.; Davison, A.; Katcher, M.L.; Lichtenberg, D.W.; Merrill, R.E.; Smart, J.C. J. Organomet. Chem.<br />
1971, 27, 271; b) Knapp, R. ; Rehahn, M. J. Organomet. Chem. 1993, 452, 235-40<br />
18b<br />
OH<br />
B<br />
OH<br />
OH<br />
B<br />
OH<br />
21
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
19 +<br />
[Pd]<br />
2 R-PTZ-X (R-PTZ) n Fc Fc-PTZ-Fc<br />
+<br />
where: Fc = Fe PTZ =<br />
20-25 26<br />
Scheme 9<br />
N<br />
S<br />
20 : R = Br, n = 1<br />
21 : R = Br, n = 2<br />
22 : R = H, n = 1<br />
23 : R = H, n = 2<br />
24 : R = I, n = 1<br />
25 : R = I, n = 2<br />
+ oligomers<br />
X = Br, I<br />
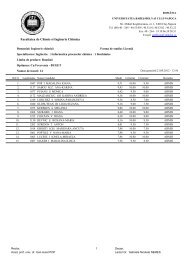
Table 1. The Negishi cross-coupling syntheses data; reagents, products, yields *<br />
Entry [Pd] R X<br />
Products<br />
Obs.<br />
Catalyst<br />
Fc(PTZ-R)2 Fc-PTZ-R Fc-PTZ-Fc<br />
1 Pd[(P<strong>Ph</strong>3)4]<br />
0.1%<br />
H Br 9.4 11 - r.t., 3 days<br />
2 Pd[(P<strong>Ph</strong>3)4]<br />
0.2%<br />
Br I 2.8 21.9 - r.t., 3 days<br />
3 Pd[(P<strong>Ph</strong>3)4] Br Br 6.6 12.4 - r.t., 3 days<br />
2.1%<br />
(9.6) (18.6)<br />
4 PdCl2(P<strong>Ph</strong>3)2 Br Br 8.2 11.0 - r.t., 3 days<br />
5%<br />
(11.2) (15.1)<br />
5 PdCl2(P<strong>Ph</strong>3)2 Br I 21.6 31.1 - r.t., 3 days<br />
3.9%<br />
(24.3) (42.4)<br />
6 Pd[(P<strong>Ph</strong>3)4]<br />
5%<br />
I I 1 9.8 3 r.t., 4 days<br />
7 Pd[(P<strong>Ph</strong>3)4]<br />
5%<br />
I I 6 2 4 r.t., 5 days<br />
8. Pd[(P<strong>Ph</strong>3)4] Br Br 28.1 12.8 - r.t., 5 days<br />
2.5%<br />
(31.2) (14.2)<br />
9. Pd[(P<strong>Ph</strong>3)4] Br Br 15.3 11.8 - 1 hr. reflux,<br />
2.5%<br />
2 days r.t.<br />
* Yields are reported to ferrocene; in brackets are presented yields calculated after subtraction of<br />
the recovered ferrocene<br />
Several attempts have been made to synthesize the desired macrocycle 27 directly from<br />
19 and 4b using 1:1 stoichiometry and high dilution technique (Scheme 15). Unfortunately, the<br />
isolated products were the corresponding dyads and triads (24-26), as well as some oligomers<br />
(identified by MS-FAB+). In one case it was isolated a compound in a very low yield which<br />
presents in mass spectra (ESI, FAB+) a peak corresponding to the molecular mass of the target<br />
macrocycle (930).<br />
19 + 4b<br />
PdCl 2 (P<strong>Ph</strong> 3 ) 2<br />
r.t, 7-10 days<br />
Fe<br />
Scheme 10<br />
N<br />
S<br />
S<br />
N<br />
27<br />
Fe<br />
22
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
This compound has a pronounced tendency to oxidize, revealed by the FAB+ technique<br />
(in the spectrum appears a significant peak at M + 16). Unfortunately, due to the very small<br />
amount and to the fact that the purity of the compound was not high, the NMR spectrum is<br />
ambiguous and could not confirm the assigned macrocyclic-type structure.<br />
2D-NMR experiments were performed in order to figure out the conformation adopted by<br />
triads in solution. The most useful experiment is NOESY. Analyzing the spectra, could not been<br />
identified cross-peaks between H 1 and H 4 , or H 6 and H 9 , respectively. This means that in solution<br />
the triads are conformationally free, the electronic interactions between the two phenothiazine<br />
units being too weak at room temperature in solution to prevent the free rotation of the ferrocene<br />
core, having the iron as a pivot (Scheme 11).<br />
R<br />
R<br />
H6 H6 H 9<br />
H9<br />
Hex<br />
N<br />
S<br />
H4 H4 S<br />
N<br />
Hex<br />
H 1<br />
H 1<br />
Fe<br />
R<br />
Hex<br />
N<br />
S<br />
Fe<br />
Scheme 11<br />
S<br />
N<br />
Hex<br />
R<br />
R<br />
R<br />
S<br />
N<br />
Hex<br />
S<br />
N<br />
Hex<br />
Unfortunately, suitable crystals for X-ray structure analysis could not be grown for these<br />
triads. Nevertheless, the dyads 20 and 22 furnished good crystals for analysis. ORTEP drawings<br />
are depicted in Figure 1.<br />
a b<br />
Figure 1. ORTEP drawings of compounds 20 (a) and 22 (b).<br />
Synthesis of macrocycles using the McMurry reductive coupling of carbonyl compounds<br />
Another considered way for the synthesis of macrocycles is to convert the triad 21 into<br />
the bis-formyl derivative and to close the macrocycle using the McMurry procedure for<br />
transformation of aldehydes into alkenes.<br />
The conversion of bromine-dyad 20, and dibromine-triad 21, respectively, was achieved<br />
in good yields and on a gram scale using a lithium-bromine exchange and a subsequent<br />
quenching of the lithiated species with dry DMF (Scheme 12).<br />
(Br-PTZ) n Fc<br />
20 : n = 1<br />
21 : n = 2<br />
where: Fc = Fe PTZ =<br />
Scheme 12<br />
(OHC-PTZ) n Fc<br />
28 : n = 1; yield 80%<br />
29 : n = 2; yield 65%<br />
N<br />
S<br />
Fe<br />
23
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
The formation of the formyl intermediates was proved by the MS spectra, in which occur<br />
the corresponding molecular peaks. Also, the 1 H-NMR spectra present the characteristic singlet<br />
signal of the formyl groups (28: δ 3-CHO = 9.79 ppm, 1H; 29: δ 3-CHO = 9.73 ppm, 2H).<br />
The triad 29 furnished crystals suitable to X-ray diffraction analysis. In Figure 9 two<br />
ORTEP drawings of 29 are presented. The two phenothiazine units are parallel and, surprisingly,<br />
they adopted a syn orientation. The two cyclopentadienyl cycles are almost parallel and eclipsed.<br />
a b<br />
Figure 2. ORTEP drawings of compound 29, different views<br />
We have chosen to test if the common reagent (TiCl4-Zn/Cu) is effective for our<br />
substrates. Thus, we have attempted the McMurry coupling of 3-ferrocenyl-7-formyl-10-nhexyl-10H-phenothiazine<br />
(28, Scheme 13) using the Mukaiyama’s procedure 25 and high dilution<br />
conditions.<br />
2<br />
OHC<br />
N<br />
S<br />
Fe<br />
[Ti]/THF, Py<br />
48 hrs., reflux<br />
60%<br />
28 30<br />
Scheme 13<br />
Fe<br />
The separation of the products by silica gel column chromatography gave the desired<br />
bis(ferrocenyl-phenothiazinyl)ethene (30) in 60% yield and a few amount of the reduced species<br />
of the starting material.<br />
This result prompted us to subject 29 to a reductive coupling reaction in the conditions<br />
mentioned above, which, after work-up, afforded the desired macrocyclic product 31 (Scheme<br />
14) in an excellent yield (73%). This excellent yield for a macrocyclization reveals the efficient<br />
template effect of the titanium low-valent species.<br />
OHC<br />
OHC<br />
N<br />
S<br />
S<br />
N<br />
29<br />
Fe<br />
[Ti]/THF, Py<br />
reflux, 48 hrs<br />
73.2%<br />
Scheme 14<br />
The 1 H-NMR spectrum of 31 presents, beside the signals characteristic to the two n-hexyl<br />
groups and for the 1, 1’-disubstituted ferrocene, in the aromatic region the signals corresponding<br />
S<br />
N<br />
Hex<br />
S<br />
N<br />
Hex<br />
N<br />
S<br />
31<br />
N<br />
S<br />
Fe<br />
Fe<br />
24
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
to the two magnetic equivalent phenothiazine units, and the vinylic protons as a singlet (δ = 6.55<br />
ppm) (Figure 3).<br />
7.5<br />
7.0<br />
6.5<br />
Figure 3. The 1 H-NMR spectrum of 31 (CDCl3, 300 MHz, r.t.)<br />
6.0<br />
5.5<br />
5.0<br />
4.5<br />
The mass spectrum is in accordance with the assigned structure.<br />
The X-rays analysis performed on monocrystal showed us that the vinylene bridge<br />
presents the cis configuration, and that the two phenothiazine units lie in an anti orientation.<br />
These phenothiazine units are a little beat twisted from a superposed arrangement, which is<br />
preferred by the bis(phenothiazinyl)ferrocene derivatives (Figure 4). This fact suggests an<br />
electronic communication between the two phenothiazines through the vinylene bridge. The<br />
other atropisomer (with a syn orientation of the phenothiazinic units) could not been detected by<br />
means of NMR spectrometry. Also, whilst there are described ferrocene macrocycles formed by<br />
McMurry reductive coupling with the resulted vinylene bridge having both cis and trans<br />
configurations, the trans isomer of 31 was not isolated.<br />
a b<br />
Figure 4. The ORTEP drawing of 31: a) view along a perpendicular axis to the cyclopentadienyl rings; b)<br />
view along a parallel axis with the cyclopentadienyl rings<br />
4.0<br />
3.5<br />
3.0<br />
2.5<br />
2.0<br />
1.5<br />
1.0<br />
0.5<br />
25
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
In order to suppress the electronic communication through the vinylene bridge, this was<br />
catalytically reduced with molecular hydrogen in 1,2-dichloroetane to an ethylene bridge<br />
(Scheme 15).<br />
N<br />
S<br />
S<br />
N<br />
Fe<br />
H 2 /10% Pd/C<br />
r.t., 48 hrs.<br />
98%<br />
31 32<br />
Scheme 15<br />
The signals of the phenothiazinic protons in the 1 H-NMR spectrum of 32 are slightly<br />
shifted up-field compared with the same protons of the 31. Also, the spectrum presents the signal<br />
of the ethylene bridge (δ -CH2-CH2- = 2.71 ppm) as a singlet. Unfortunately, this compound<br />
crystallizes in sharp needles, which are unsuitable for X-rays diffraction analysis. However, the<br />
mass spectrum (FAB+) confirms the assign structure.<br />
<strong>Ph</strong>isyco-Chemical Properties of the Synthesized Dyads, Triads and Macrocycles<br />
The dyads, triads, and the macrocycles were analyzed using cyclic voltammetry.<br />
The results are presented in the Table 2. Due to the presence of two redox active units (ferrocene<br />
and phenothiazine, respectively) our expectation was to find two oxidation waves. Indeed, the<br />
cyclo-voltammograms of the dyads show two oxidation steps for the ferrocene and the<br />
phenothiazine unit, respectively. The first step (0.57 V) is due to the ferrocene moiety, and the<br />
second (1.01-1.02 V) to the phenothiazine rest, respectively (see Table 5, entry 1). In the case of<br />
the bis(phenothiazinyl)ferrocene triads there are three redox waves (see Table 5, entry 2) despite<br />
the fact that the molecules are symmetric. This fact can be a consequence of the electronic<br />
communication between the two redox active phenothiazine centres. The electronic<br />
communication can take place through the already oxidized ferrocene or through space by πstacking.<br />
It was previously shown by the X-rays diffraction that in the solid state the two<br />
phenothiazine moieties are superposed, most probably due to the π-stacking. In the case of<br />
bis(ferrocenyl)-phenothiazine, two redox steps occur in the voltammogram, one due to the two<br />
ferrocene units (two electrons process) and one (one electron process). This means that the two<br />
feerocene are independent each other and, therefore, have the same redox potential.<br />
One reason for the synthesis of the target macrocycles is to investigate this electronic<br />
communication in solution because the two phenothiazine units are constrain at least partly to<br />
superpose. The cyclo-voltammograms present the same electronic behavior like the triads. The<br />
only difference in that the first oxidation step assigned to the phenothiazine units are at lower<br />
potentials in the case of macrocycles. The gap between the oxidation steps of the phenothiazines<br />
in the case of the non-cyclic triads is lower (0.111-0.169 V) than in the case of the macrocycles<br />
(0.198 V for 31 and 0.238 V for 32, respectively). This fact can be take as a measure of<br />
electronic communication (one phenothiazine unit playing the role of a rich-electron substituent<br />
of the phenothiazine which oxidizes first) in the way that the greater the gap, the stronger the<br />
communication. Not surprisingly, the strongest electronic communication is found in the<br />
macrocyclic triads, especially for 32 in which the ethylene bridge is flexible (compared with the<br />
vinylene bridge of the 31) and short enough to constrain the superposition of the phenothiazine<br />
units in solution. This superposition is less probable in solution for the non-cyclic triads due to<br />
the solvation of the molecule and to the free rotation around the ferrocene, respectively.<br />
N<br />
S<br />
S<br />
N<br />
Fe<br />
26
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
Table 2. Cyclic voltammetry data of some ferrocene-phenothiazine derivatives<br />
Entry Cyclic Voltammogram E1/2 (V) Compound<br />
1<br />
2<br />
3<br />
4<br />
5<br />
I (µA)<br />
I (µA)<br />
I (µA)<br />
I (µA)<br />
I (µA)<br />
4<br />
2<br />
0<br />
-2<br />
-4<br />
-6<br />
RG 27 a<br />
100 mV/s, CH2Cl2<br />
-8<br />
1,4 1,2 1,0 0,8 0,6 0,4 0,2 0,0 -0,2 -0,4 -0,6<br />
4<br />
2<br />
0<br />
-2<br />
-4<br />
E (V)<br />
-6<br />
1,5 1,0 0,5 0,0 -0,5<br />
3<br />
2<br />
1<br />
0<br />
-1<br />
-2<br />
-3<br />
RG 27 b<br />
100 mV/s<br />
E (V)<br />
-4<br />
1,4 1,2 1,0 0,8 0,6 0,4 0,2 0,0 -0,2 -0,4 -0,6<br />
2,0<br />
1,5<br />
1,0<br />
0,5<br />
0,0<br />
-0,5<br />
-1,0<br />
-1,5<br />
-2,0<br />
-2,5<br />
2<br />
1<br />
0<br />
-1<br />
-2<br />
RG 19 c<br />
CH2Cl2, 100 mV/s<br />
E (V)<br />
RG 68<br />
CH 2 Cl 2 , 100 mV/s<br />
1,4 1,2 1,0 0,8 0,6 0,4 0,2 0,0<br />
E (V)<br />
RG 73<br />
CH2Cl2, 100 mV/s<br />
-3<br />
1,4 1,2 1,0 0,8 0,6 0,4 0,2 0,0<br />
2.4. SHAPE-PERSISTENT MACROCYCLES<br />
E (V)<br />
0.572<br />
1.020<br />
0.549<br />
0.946<br />
1.115<br />
0.576<br />
1.042<br />
0.563<br />
0.904<br />
1.102<br />
0.521<br />
0.798<br />
1.036<br />
Br<br />
Br<br />
Br<br />
N<br />
S<br />
N<br />
S<br />
S<br />
N<br />
Fe<br />
Fe<br />
Fe Fe<br />
A. Introduction<br />
In the last decade a lot of shape-persistent macrocycles on the nanometre scale have been<br />
synthesized and characterized. Due to a) the conformational rigidity, and b) to the fact that the<br />
interior space is separated by the exterior these macrocycles were good candidates for various<br />
supramolecular studies, such as organisation into and transport through porous molecular<br />
crystals 44,45 or pattern formation at interfaces. 46 The basic structural type is represented by the<br />
44 a) Henze, O.; Lenz, D.; Schäfer, A.; Franke, P.; Schlüter, A.D. Chem. Eur. J. 2002, 357-365; b) Werz, T.B.;<br />
Staeb, T.H.; Benisch, C.; Rausch, B.J.; Rominger, F.; Gleiter, R. Org. Lett. 2002, 4, 339-342; c) Campbell, K.;<br />
Kuehl, C.J.; Ferguson, M.J.; Stang, P.J.; Tykwinski, R.R. J. Am. Chem. Soc. 2002, 124, 7266-7267.<br />
45 See, for example: Venkataraman, D.; Lee, S.; Zhang, J.; Moore, J.S. Nature 1994, 371, 591-593.<br />
46 See, for example: a) Borissov, D.; Ziegler, A.; Höger, S.; Freyland, W. Langmuir 2004, 20, 2781-2784; b) Grave,<br />
C.; Schlüter, A.D. Eur. J. Org. Chem. 2002, 3075-3098; c) Höger, S.; Bonrad, K.; Mouran, A.; Beginn, U.; Möller,<br />
M. J. Am. Chem. Soc. 2001, 123, 5651-5659; d) Zhao, D.; Moore, J.S. Chem. Commun. 2003, 807-818.<br />
N<br />
S<br />
S<br />
N<br />
N<br />
S<br />
S<br />
N<br />
S<br />
N<br />
Fe<br />
Fe<br />
27
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
phenylene-acetylene macrocycles, 47 which differ in the ring size (up to 5.5 nm) and in the sidechain<br />
pattern. Several macrocyclic phenylacetylenes have been shown to have interesting<br />
properties, such as self-association in solution, 48 the existence of liquid crystaline phases, 49 or the<br />
capability to bind small molecules. 50<br />
B. Synthesis of the Intermediates<br />
The strategy is based on the sequential Sonogashira coupling reactions between dihalophenothiazines<br />
and bis(ethynyl)-phenothiazine, which could be readily available from dihalophenothiazines.<br />
The synthesis of bis(ethynyl)-phenothiazine (33) was previously described using<br />
several ways: by Sonogashira coupling of dibromo-phenothiazine with trimethylsilyl-acetylene<br />
(TMS-acetylene), by converting formyl-phenothiazines using Corey-Fuchs protocol or,<br />
alternatively, the Ohira-Bestmann transformation. 51<br />
It is well known that iodine derivatives react under mild conditions and easier than<br />
bromine ones. As a consequence, we have performed this Sonogashira coupling using diiodophenothiazine<br />
(Scheme 16); the reaction underwent smoothly, at room temperature, to afford the<br />
bis(TMS-ethynyl)-phenothiazine (34) in 95% yield; the deprotection of TMS-groups was<br />
achieved with TBAF/CH2Cl2.<br />
I<br />
N<br />
S<br />
4b<br />
34<br />
I<br />
+ 2<br />
TBAF / CH 2 Cl 2 + THF<br />
r.t., 90 min.<br />
95%<br />
SiMe 3<br />
4% PdCl 2 (P<strong>Ph</strong> 3 ) 2 , 8% CuI<br />
piperidine, r.t., 5 min.<br />
98%<br />
N<br />
S<br />
Me Si 3 SiMe3 34<br />
N<br />
S<br />
33<br />
Scheme 16<br />
The 1 H-NMR of 33 presents the signal corresponding to the two 1-alkynyl protons (δ = 3.03<br />
ppm).<br />
The next step was the synthesis of the first precursor (35). The Sonogashira coupling<br />
using the previously described conditions and the diiodo-phenothiazine 4b in 50% excess<br />
(Scheme 17) afforded the diiodo-diethynylene-bridged phenothiazine triad 35 in fair yield (55%)<br />
and some other products, most probably oligomers with 4 or more phenothiazine units.<br />
4b + 33<br />
4% PdCl 2 (P<strong>Ph</strong> 3 ) 2 , 8% CuI<br />
piperidine, r.t., 5 hrs.<br />
55%<br />
N<br />
I<br />
S<br />
Scheme 17<br />
47<br />
Zhang, J.; Pesak, D.J.; Ludwick, J.L.; Moore, J.S.; J. Am. Chem. Soc. 1994, 116, 4227-4239.<br />
48<br />
a) Shetty, A.S.; Zhang, J.; Moore, J.S. J. Am. Chem. Soc. 1996, 118, 1019-; b) Tobe, Y.; Nagano, A.; Kawabata,<br />
K.; Sonoda, M.; Naemura, K. Org. Lett. 2000, 2, 3265-, and references cited therein.<br />
49<br />
a) Zhang, J.; Moore, J.S. J. Am. Chem. Soc. 1994, 116, 2655; b) Collins, S.K.; Yap, G.P.A.; Fallis, A.G. Org. Lett.<br />
2000, 2, 3189-.<br />
50<br />
Morrison, D.L.; Höger, S. Chem. Commun. 1996, 2313-14.<br />
51<br />
Krämer, C.S.; Zeitler, K.; Müller, T.J.J. Org. Lett. 2000, 2(23), 3723-3726, and references cited therein.<br />
N<br />
S<br />
35<br />
S<br />
N<br />
I<br />
28
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
In order to reduce the ratio of oligomerization, we have tested the chemoselectivity of the<br />
Sonogashira coupling using bromo-iodo-phenothiazine 5 with the selective obtaining of the<br />
dibromo-analog of 35 (36, Scheme 18).<br />
5 + 33<br />
4% PdCl 2 (P<strong>Ph</strong> 3 ) 2 , 8% CuI<br />
piperidine, r.t., 24 hrs.<br />
N<br />
Br<br />
S<br />
Scheme 18<br />
The yield was higher (72%), and the selectivity was high, and, as in the Negishi or<br />
Suzuki coupling involving bromo-iodo-phenothiazine, only traces of the bromine-iodineethynylene-bridged<br />
phenothiazine triad have been identified by mass spectrometry. Another<br />
proof of the selective cross-coupling was brought by the 13 C-NMR spectrum of 36, in which does<br />
not appear a signal corresponding to a Csp2-I carbon atom (signal at about δ 84-85 ppm).<br />
The second precursor has to have two terminal ethynyl groups; the synthesis of 37 was<br />
performed by a Sonogashira coupling between 35 and TMS-acetylene (Scheme 19), and a<br />
subsequent deprotection of the TMS-protected intermediate 38 with TBAF.<br />
35<br />
+<br />
SiMe 3<br />
4% PdCl 2 (P<strong>Ph</strong> 3 ) 2 , 8% CuI<br />
piperidine, r.t., 1 hr.<br />
38<br />
TBAF/CH 2 Cl 2<br />
r.t., 1 hr.<br />
N<br />
Scheme 19<br />
In the 1 H-NMR spectrum of 37 the signal corresponding to the two terminal ethynyl<br />
protons appears at 3.05 ppm.<br />
C. The Cyclization Attempt<br />
The cyclization was performed using the high dilution technique. An equimolar mixture<br />
of the two precursors (35 and 37, respectively) was subjected to the Sonogashira cross coupling<br />
reaction (Scheme 20). The 1 H-NMR spectrum of the main product is complex. In the aliphatic<br />
region there are many signals due to at least two magnetically non-equivalent n-hexyl residues,<br />
which is different as I would expect taking into consideration the 6 symmetrical substituted<br />
phenothiazines from the target structure of the macrocycle. Also, these signals are shifted<br />
downfield compared with those assigned to an n-hexyl core from a non-cyclic n-hexyl-10Hphenothiazine<br />
derivative. In the aromatic region, the signals are spread between 5.5 and 9.5 ppm.<br />
The spectrum of the sample extracted with THF looks more familiar with a spectrum of an nhexyl-10H-phenothiazine<br />
derivative; however, in the aliphatic region the situation is the same as<br />
previously described. In the aromatic region the signals are shifted downfield compared with the<br />
precursors, and are more than 3 as I would expect for a symmetrical structure. This behavior can<br />
be due to the free rotation of the phenothiazine units having the ethynyl units as pivots. In this<br />
situation, there are two kinds of phenothiazine units: some with the N-n-hexyl rest oriented<br />
outside the cycle, and the other having the N-n-hexyl rest oriented inside.<br />
S<br />
N<br />
S<br />
36<br />
N<br />
S<br />
37<br />
S<br />
N<br />
Br<br />
S<br />
N<br />
29
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
35 +<br />
37<br />
4% PdCl 2 (P<strong>Ph</strong> 3 ) 2 , 8% CuI<br />
piperidine/THF, r.t., 48 hrs.<br />
N<br />
N<br />
S<br />
S<br />
Scheme 20<br />
Mass spectrometry experiments were performed in order to confirm the macrocyclic<br />
structure. ESI-MS experiments have failed, whilst the MALDI gave a very small signal at<br />
m/e=1831 (for the crude precipitate) and at m/e=1832 (for the THF extract). These values are in<br />
the accepted range of measuring errors (± 0.1%, M=1830) and have shown the existence of the<br />
target macrocycle.<br />
In order to determine the electrochemical properties of the (poly)ethynylene triads, 35<br />
was subjected to cyclic voltammetry determination. The results are presented in Table 5.<br />
The cyclic-voltammogram of 35 presents one two-electrons oxidation step (0.92 V)<br />
assigned to the two marginal phenothiazine units and an one-electron oxidation step (1.02 V)<br />
corresponding to the internal phenothiazine moiety. The two-electron oxidation step shows that<br />
the marginal phenothiazine units are not electronically connected and they are oxidizing<br />
independent at the same potential.<br />
Table 5. Redox properties and the cyclic-voltammogram of 35<br />
CV Graph Oxidation step E1/2 (V) Structure<br />
I (µA)<br />
15<br />
10<br />
5<br />
0<br />
-5<br />
-10<br />
-15<br />
RG 69<br />
500 mV/s<br />
1,4 1,2 1,0 0,8 0,6 0,4 0,2 0,0<br />
E (V)<br />
2.5. PHENOTHIAZINE CORON<strong>AND</strong>S<br />
1 0.924<br />
2 1.020<br />
A. The Synthesis of the Intermediates<br />
The conversion of 1 into 3,7-bis(hydroxymethyl)-10-ethyl-10H-phenothiazine (39) was<br />
achieved in two steps. The first one has involved a Br-Li exchange with nBuLi, followed by the<br />
addition of DMF as electrophile. The work-up with 5% HClaq afforded the diformyl-derivative<br />
40 (Scheme 21). A subsequent reduction of 40 in mild conditions gave the first key intermediate<br />
39.<br />
N<br />
S<br />
S<br />
N<br />
38<br />
N<br />
I<br />
S<br />
S<br />
S<br />
N<br />
N<br />
N<br />
S<br />
S<br />
N<br />
I<br />
30
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
N<br />
1° 2.5 eq. nBuLi/THF/-78°C<br />
Br S Br 2° 2.5 eq. dry DMF/-78°C<br />
OHC S CHO 10 min., r.t. H C S CH<br />
3° 5% HCl 2<br />
2<br />
aq<br />
OH OH<br />
1a<br />
65-71%<br />
40<br />
99%<br />
39<br />
N<br />
Scheme 21<br />
NaBH 4 /i-propanol<br />
The 1 H-NMR spectrum of 39 presents the three sets of signals in the aromatic zone, fact which<br />
signifies the symmetrical substitution of the molecule. Besides of these, the signal corresponding<br />
to the hydroxymethyl substituents appears in the spectrum (δ CH2 = 4.37 ppm, δ OH = 5.10 ppm,<br />
Figure 14). The signals of the methylene and the hydroxyl of the hydroxymethyl groups are<br />
splitted due to their coupling ( 3 J = 5.70 Hz).<br />
The synthesis of the other key intermediate of our strategy also started from 1a through a<br />
Br-Li exchange with nBuLi. In this case, the electrophile was trimethylborate, followed by the<br />
addition of pinacole in order to generate the stable phenothiazine bis(boronic ester) (Scheme 22).<br />
N<br />
Br S<br />
Br<br />
1a<br />
1° 2.5 eq. nBuLi/THF/-78°C<br />
2° 2.5 eq. dry B(OMe) 3 /-78°C<br />
3° 2.5 eq. dry pinacole, r.t.<br />
50-53%<br />
Scheme 22<br />
The Suzuki cross-couplings of the n-hexyl-analog of 41 were presented above. In this<br />
strategy two other coupling partners were considered: bromo-benzenes m-substituted with OH<br />
and NH2, respectively. The synthesis of the ditopic intermediates for macrocyclization was<br />
realized using standard conditions for the cross-coupling: Pd[P<strong>Ph</strong>3]4 as catalyst, monoglym or<br />
diglym/water, and an inorganic carbonate (Scheme 23). The column chromatography<br />
purification (petroleum ether:ethyl acetate = 3:2) afforded the desired products 42 and 43, as well<br />
as some small amounts of mono-substituted by-products.<br />
O<br />
B<br />
O<br />
N<br />
S<br />
41<br />
O<br />
B<br />
O<br />
+<br />
Br<br />
O<br />
B<br />
O<br />
N<br />
S<br />
41<br />
O<br />
B<br />
O<br />
diglym:water = 2:1<br />
Y reflux, overnight<br />
S<br />
Scheme 23<br />
5% Pd[P<strong>Ph</strong> 3 ] 4 , K 2 CO 3<br />
N<br />
Y<br />
42: Y = OH , yield 60%<br />
43: Y = NH2 , yield 65%<br />
Y<br />
The mass spectra of the products present the molecular peak (42: M/z = 613, 43: M/z =<br />
611). The 1 H-NMR spectrum of 42 presents in the aromatic region partial overlapped 7 signals<br />
corresponding to 14 protons. Also, the phenolic hydroxyl protons give in the spectrum a signal<br />
having the integral for two protons. Also, the 13 C-NMR spectrum confirms the symmetrical<br />
substitution of the phenothiazine core, due to the fact that in the aromatic region appear 12<br />
signals corresponding to the 12 types of carbon atoms of the molecule.<br />
The NMR spectra of 43 are almost similar to those of 42, with the difference that in the<br />
1<br />
H-NMR spectrum of 43 appears the signal corresponding to the 2 magnetic equivalent amino<br />
groups (δ NH2 = 5.17 ppm).<br />
B. Synthesis of Coronands Having <strong>Ph</strong>enothiazine Units<br />
The ditosylated pentaethyleneglycols were prepared by tosylation of the corresponding<br />
polyethylene glycols (Scheme 24).<br />
N<br />
31
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
HO<br />
O n<br />
H<br />
2.2 equiv. TsCl<br />
NaOH/THF<br />
2 hrs., 0-5°C<br />
Scheme 24<br />
The ditosyl derivatives were analyzed by NMR spctroscopy.<br />
TsO<br />
n = 1-5<br />
ATTEMPT OF MACROCYCLIZATION OF 39<br />
One attempt of the synthesis of phenothiazine-containing macrocycle starting from 39<br />
was made using ditosylated triethyleneglycole (Scheme 25).<br />
N<br />
H2C S<br />
CH2 OH 39 OH<br />
N<br />
O n<br />
H2C S<br />
CH2 O O O O<br />
Conditions: 1° 2 eq. NaH/THF/reflux 1 hr.<br />
2° 1 eq. diTs triethyleneglycole over 6 hrs/THF/reflux 48 hrs.<br />
Scheme 25<br />
39 was converted into the disodium salt in order to increase its reactivity. After the workup<br />
of the reaction mixture, the majority of the fr1 was recovered, as well as small amounts of<br />
formyl- and diformyl-10-ethyl-phenothiazines. The same failure of the macrocyclization of a<br />
bis(hydroxymethyl)-phenothiazine derivative was reported by Bauer et al. 11<br />
MACROCYCLIZATION REACTION OF 42<br />
42 was subjected to macrocyclization with ditosylated pentaethylene glycole in<br />
acetonitrile using the high-dillution technique (Scheme 36). Cs2CO3 in excess was used as base,<br />
as well as template. The reaction afforded the target macrocycle in good yield (52%).<br />
TsO<br />
N<br />
S<br />
OH 42<br />
OH<br />
O<br />
O<br />
+<br />
O<br />
O<br />
OTs<br />
5 equiv. Cs 2 CO 3 , CH 3 CN<br />
7 days, reflux<br />
52%<br />
Scheme 26<br />
O O<br />
N<br />
S<br />
O<br />
Ts<br />
O O<br />
The product was investigated by MS, NMR and X-ray diffraction. The EI-MS spectrum<br />
presents the molecular peak (M/z = 613). One interesting feature of this macrocyclization is that<br />
after the column chromatography purification no cyclic oligomers were separated.<br />
The 1 H-NMR spectrum presents the peak distribution of the phenothiazine precursor,<br />
and, in addition, the signals characteristic to the pentaethylene glycol chain (protons c,d,e in<br />
Figure 5).<br />
44<br />
O<br />
32
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
7.5<br />
7.0<br />
6.5<br />
5'<br />
4'<br />
c<br />
6'<br />
6.0<br />
8<br />
2'<br />
b<br />
a<br />
CH3 H C 2<br />
9<br />
1<br />
N<br />
6<br />
5.5<br />
S<br />
O O<br />
d<br />
O O O O<br />
d<br />
e e e e e e<br />
Figure 5. 1 H-NMR spectrum of 44 (CDCl3, 300 MHz, r.t.)<br />
4<br />
2<br />
2'<br />
5.0<br />
6'<br />
c<br />
5'<br />
4'<br />
c<br />
4.5<br />
d<br />
a b<br />
In the aromatic area the signals were assigned studying the multiplicity, the shape and the<br />
coupling constants (Figure 6).<br />
H4, H6<br />
7.50<br />
20 20<br />
20<br />
H2, H8 H5'<br />
7.40<br />
CDCl 3<br />
7.30<br />
H2', H6'<br />
7.20<br />
41<br />
4.0<br />
5'<br />
4'<br />
c<br />
7.10<br />
6'<br />
8<br />
e<br />
3.5<br />
6<br />
S<br />
7.00<br />
3.0<br />
b<br />
a<br />
CH3 H2C 9<br />
1<br />
N<br />
O O<br />
d<br />
2'<br />
O O O O<br />
d<br />
e e e e e e<br />
H1, H9<br />
4<br />
2<br />
2'<br />
6'<br />
2.5<br />
c<br />
5'<br />
4'<br />
6.90<br />
2.0<br />
18 18<br />
Figure 6. Detail with the aromatic zone of the 1 H-NMR spectrum of 44 (CDCl3, 300 MHz, r.t.)<br />
The solid state molecular structure of 44 was determined by X-rays diffraction analysis of<br />
a monocrystal. In Figure 7 is presented the ORTEP drawing of one molecule.<br />
H4'<br />
1.5<br />
6.80<br />
33
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
Figure 7. ORTEP drawing of the X-ray diffraction analysis of a monocrystal of 44<br />
The dihedral angle between the two phenylenes of the phenothiazine core is 133.26°. The<br />
distance between the centroids of the two benzene units is 11.97Å, while the distance between<br />
the sulfur atom and the oxygen atom from the middle of the penta(oxaethylene) chain is 4.97Å.<br />
Taking into consideration that the chain is a little beat twisted, we can assume that the cavity of<br />
the macrocycle 44 has an elliptic shape, with one dimension being the double of the small<br />
dimension.<br />
The unit cell contains four macrocycles (Figure 8), having the (oxaethylene) chains<br />
oriented outside.<br />
Figure 8. Mercury drawing showing the packing of 44 in monocrystal (“wireframe” style)<br />
The packing of these four-macrocycles units generates “empty” rectangle channels (Figure 20).<br />
Two opposite faces of these columns have phenothiazines as “wall tiles”, whilst the other two<br />
opposite faces are delimited by (ethyleneoxa) chains. The distance between two sulphur atoms<br />
belonging to opposite faces is 11.98Å, and the shortest distance between two opposite ethylene<br />
units is 16.22Å.<br />
CONCLUSIONS<br />
The structural analysis of new spiro and dispiro-4[H]-1,3-dioxine derivatives carried out<br />
using NMR spectra and X-ray diffractometry revealed flexible and semiflexible structures and<br />
34
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
the cis, trans (compounds 3-8) and the like, unlike (compound 7) isomerism of these derivatives.<br />
The cyclohexane ring prefers the chair conformation, while the 4[H]-1,3-dioxine rings have<br />
chiral half-chair conformations. The investigation of the lattice of 7 showed important stacking<br />
interactions.<br />
The synthesis by an original procedure of a series of new (poly)spiranes containing 1,2dithiolane<br />
rings as cyclic disulfides leads to compounds with flexible or semi-flexible<br />
conformational behaviour, starting from readily available raw materials. The adsorption of some<br />
of these derivatives on a gold surface and the structural behaviour of the resultant 2D-SAMs<br />
were investigated using computational chemistry modelling as well as IR spectroscopy. Despite<br />
the fact that they all appear to adopt a tilted orientation and do not form a well ordered<br />
monolayer, the bidentate cyclic disulfide 32 revealed the possibility to react with both 1,2dithiolane<br />
units, giving rise to a rigid bridge-like conformation. These cyclic disulfides were<br />
effective also in the surface functionalization of gold nanoparticles. These assemblies were<br />
investigated by means of UV-Vis spectroscopy, transmission electron microscopy, and cyclic<br />
voltammetry. Again, 32 has shown interesting properties in functionalization of GNPs (provokes<br />
high aggregation degrees) and in functionalization of gold electrodes with GNPs (the<br />
functionalized electrode with 32 and GNP increased two times the current in cyclic voltammetry,<br />
compared with the bare gold electrode).<br />
During the work in the phenothiazine field, the obtaining of conjugated phenothiazinecontaining<br />
macrocycles was analyzed and pursued.<br />
First, some new valuable iodo-phenothiazine intermediates were obtained and tested in<br />
palladium catalyzed cross-coupling reactions. Despite the difficulties in coupling two richelectron<br />
aromatic systems, the results obtained using the palladium catalyzed cross-couplings,<br />
especially the Negishi coupling involving ferrocene-zincchloride derivatives as intermediates,<br />
opened us the way to achieve the synthesis of phenothiazine-ferrocene containing macrocycles.<br />
Several ferrocene-phenothiazine dyads, triads were synthesized and duly characterized, and some<br />
non-cyclic oligomers by-products were identified. The attempts to synthesize the full-conjugated<br />
bis(ferrocene-phenothiazine) macrocycle have lead to complex mixtures. The ESI-MS<br />
experiments have revealed some small amounts of the desired macrocycle (m/e=930);<br />
unfortunately, this compound could not been isolated in order to perform a full characterization.<br />
The target ferrocene-phenothiazine macrocycle possessing a vinylene bridge was<br />
obtained in good yield using a McMurry reductive coupling of the bis(formyl)ferrocenephenothiazine<br />
triad. The vinylene bridge was selective reduced to an ethylene bridge. In order to<br />
determine the redox properties, the dyads, triads and the macrocycles were subjected to cyclic<br />
voltammetry experiments. An interesting feature of the voltammograms was the non-equivalence<br />
of the phenothiazine moieties in the triads and macrocycles, proving an electronic<br />
communication between these two units. This communication could be established through the<br />
oxidized iron of ferrocene rest or through space, by π-stacking. However, the π-stacking proved<br />
to be important in the solid phase and in macrocycles.<br />
Another class of compounds taken into our attention was the phenothiazine-containing<br />
shape-persistent macrocycles. The synthetic strategy was built on the Sonogashira cross-coupling<br />
reaction between acetylene-derivatives and halo-phenothiazines. The precursors were<br />
synthesized and duly characterized. The macrocyclization main product presents in the MALDI-<br />
TOF measurement the assigned mass for the target macrocycle. Unfortunately, the purity control<br />
using a valuable (i.e. GPC method) and the improvement of the macrocyclization were not<br />
performed.<br />
Another target class of phenothiazine-containing macrocycle was represented by the<br />
phenothiazine-coronands. The desired macrocycles were synthesized in several steps, with the<br />
generation of a dihydroxy-intermediate as the key step. In macrocyclization were used<br />
ditosylated polyethyleneglicole of different lengths as electrophilic partners. The characterization<br />
of these macrocycles was made on the basis of MS, NMR, X-ray diffraction and cyclic<br />
voltammetry.<br />
35
Radu Gropeanu – <strong>Ph</strong>D Thesis Abstract<br />
List of Publications<br />
Published:<br />
1. Sailer, M.; Gropeanu, R.A.;. Müller, T.J.J. “PRACTICAL SYN<strong>THESIS</strong> OF IODO<br />
PHENOTHIAZINES. A FACILE ACCESS TO ELECTROPHORE BUILDING BLOCKS”<br />
Journal Of Organic Chemistry 2003, 68, 7509-12.<br />
2. Gropeanu, R.A.; Woiczechowski-Pop, A.; Tintas, M.; Turdean, R.; Grosu, I. “SYN<strong>THESIS</strong><br />
<strong>AND</strong> <strong>STEREOCHEMISTRY</strong> OF A NEW SERIES OF 2,2’-DISUBSTITUTED-5,5-<br />
BIS(BROMOMETHYL)-1,3-DIOXANES” Studia Univ. Babes-Bolyai, Ser. Chemia. 2005, L,<br />
247.<br />
Manuscripts:<br />
1. Gropeanu, R.A.; Grosu, I. “SYN<strong>THESIS</strong> <strong>AND</strong> <strong>STEREOCHEMISTRY</strong> OF SOME NEW<br />
SPIRO BENZO-1,3-DIOXANE DERIVATIVES” Central European Journal of Chemistry 2005,<br />
submitted.<br />
2. Gropeanu, R.A.; Tintas, M.; Pilon, C. ; Morin, M.; Breau, L.; Turdean, R.; Grosu, I.<br />
“SYN<strong>THESIS</strong>, <strong>STEREOCHEMISTRY</strong> <strong>AND</strong> ADSORPTION STUDIES OF NEW<br />
(POLY)SPIRANES CONTAINING 1,2-DITIOLANE UNITS “Monatshefte für Chemie 2005,<br />
submitted.<br />
Posters:<br />
1. Roiban, D.G., Gropeanu, R.A., Gâz, S.A., Grosu, I. "SPECTROSCOPIC METHODS USED<br />
FOR STRUCTURE INVESTIGATION OF SOME NEW DIOXANES DERIVATIVES", 5th<br />
European Conference on Mineralogy and Spectroscopy (ECMS) in Vienna, Austria, September<br />
4th - 8th 2004<br />
2. Gâz, S. A., Gropeanu, R.A., Roiban, D., Grosu, I. "SYN<strong>THESIS</strong>, <strong>STEREOCHEMISTRY</strong><br />
<strong>AND</strong> REACTIVITY OF SOME 2-METHYL-2-(3’-NITROPHENYL)-4-HYDROXYMETHYL-<br />
1,3-DIOXOLANE" 5th European Conference on Mineralogy and Spectroscopy (ECMS) in<br />
Vienna, Austria, September 4th - 8th 2004<br />
36