Organometallics was closed and heated at 120 °C for 20 h. Then, it was cooled to room temperature, and I n C 3 F 7 (8 μL, 0.054 mmol) was added. After 24 h, 1 H, 19 F, and 31 P{ 1 H} NMR spectra <strong>of</strong> the resulting dark red-brown solution were measured. [Rh(η 5 -Cp*)( n C 3 F 7 )I(PMe 3 )] (4f) 21 and [Rh(η 5 -Cp*)I 2 (PMe 3 )] 69,70 were the main reaction products, accounting respectively for 40% and 27% <strong>of</strong> the mixture (the ratio is based on the integration <strong>of</strong> the 31 P{ 1 H} NMR spectra <strong>of</strong> the mixture). Their NMR data were in agreement with those previously reported. 4f: 1 H NMR (300.1 MHz, C 6 D 6 ) δ 1.50 (d, 3 J RhH = 3.3 Hz, 15 H, C 5 Me 5 ), 1.24 (d, 2 J PH = 10.6 Hz, 9 H, PMe 3 ). 19 F NMR (282.4 MHz, C 6 D 6 ): δ −66.1 (AB d, 2 J FF = 272.4 Hz, 1 F, RhCF 2 ), −68.7 (AB d, 2 J FF =269.3 Hz, 1 F, RhCF 2 ), −78.5 (t, 3 J FF =11.3Hz,3F,CF 3 ), −113.3 (AB d, 2 J FF = 279.7 Hz, 1 F, CF 2 CF 3 ), −114.9 (AB d, 2 J FF = 279.1 Hz, 1 F, CF 2 CF 3 ). 31 P{ 1 H} NMR (121.5 MHz, C 6 D 6 ): δ 2.7 (dm, 1 J RhP =150.7Hz). [Rh(η 5 -Cp*)I 2 (PMe 3 )]: 1 H NMR (300.1 MHz, C 6 D 6 ): δ 1.58 (d, 3 J RhH =3.4Hz,15H,C 5 Me 5 ), 1.48 (d, 2 J PH =10.1Hz,9H,PMe 3 ). 31 P{ 1 H} NMR (121.5 MHz, C 6 D 6 ): δ −2.0 (d, 1 J RhP =138.2Hz). [Rh(η 5 -Cp*)( n C 4 F 9 )I(PMe 3 )] (4g). This was prepared from [Rh(η 5 -Cp*)(η 2 -C 2 H 4 ) 2 ] (150 mg, 0.51 mmol), PMe 3 (0.61 mmol), and I n C 4 F 9 (90 μL, 0.51 mmol) in a similar way to 3a (method A). Column chromatography (silica gel) using Et 2 O as eluent gave an orange fraction (R f = 0.95), which was evaporated to dryness to give an orange oil (40 mg, 12%). X-ray quality single crystals were obtained by slow evaporation <strong>of</strong> a toluene solution. Mp: 153−156 °C. Anal. Calcd for C 17 H 24 F 9 IPRh: C, 30.93; H, 3.66. Found: C, 31.01; H, 3.36. 1 H NMR (400.9 MHz, C 6 D 6 ): δ 1.49 (d, 4 J PH = 2.8 Hz, 15 H, C 5 Me 5 ), 1.23 (d, 2 J PH = 10.5 Hz, 9 H, PMe 3 ). 13 C{ 1 H} NMR (75.5 MHz, C 6 D 6 ): δ 101.5 (dd, 1 J RhC = 4.5 Hz, 2 J PC = 2.9 Hz, C 5 Me 5 ), 19.0 (d, 1 J PC = 33.3 Hz, PMe 3 ), 10.4 (s, C 5 Me 5 ). The signals corresponding to the carbons <strong>of</strong> the n C 4 F 9 group were not observed. 19 F NMR (188.3 MHz, C 6 D 6 ): δ −66.2 (AB d, 2 J FF = 272.1 Hz, 1 F, RhCF 2 ), −68.3 (AB d, 2 J FF = 273.0 Hz, 1 F, RhCF 2 ), −80.8 (s, 3 F, CF 3 ), −110.3 (AB d, 2 J FF = 285.4 Hz, 1 F, C β F 2 ), −111.7 (AB d, 2 J FF = 285.4 Hz, 1 F, C β F 2 ), −124.6 (m, 2 F, C γ F 2 ). 31 P{ 1 H} NMR (81.0 MHz, C 6 D 6 ): δ 2.7 (dm, 1 J RhP = 150.5 Hz). [Rh(η 5 -Cp*)(CFCF 2 )I(PMe 3 )] (4h). This was prepared from [Rh(η 5 -Cp*)(η 2 -C 2 H 4 ) 2 ] (157 mg, 0.53 mmol), PMe 3 (0.64 mmol), and ICFCF 2 (53 μL, 0.56 mmol) in a similar way to 3a (method A). Column chromatography (silica gel) using Et 2 O/n-hexane (3:1) as eluent gave an orange fraction (R f = 0.6), which was evaporated to dryness to give an orange solid (40 mg, 14%). Mp: 132−135 °C. Anal. Calcd for C 15 H 24 F 3 IPRh: C, 34.51; H, 4.63. Found: C, 34.21; H, 4.68. 1 H NMR (300.1 MHz, C 6 D 6 ): δ 1.53 (d, 4 J PH = 2.9 Hz, 15 H, C 5 Me 5 ), 1.30 (d, 2 J PH = 10.8 Hz, 9 H, PMe 3 ). 13 C{ 1 H} NMR (75.5 MHz, C 6 D 6 ): δ 160.2 (ddd, 1 J CF = 311.7 and 259.8 Hz, 2 J CF = 47.4 Hz, CF CF 2 ), 100.1 (dd, 1 J RhC = 4.5 Hz, 2 J PC = 3.0 Hz, C 5 Me 5 ), 18.3 (d, 1 J PC = 34.3 Hz, PMe 3 ), 10.0 (s, C 5 Me 5 ). The signal corresponding to the CFCF 2 carbon was not observed. 19 F NMR (282.4 MHz, C 6 D 6 ): δ −90.4 (dd, 2 J FF = 93.9 Hz, 3 J FF cis = 38.8 Hz, RhCCF trans to Rh), −121.9 (dd, 2 J FF = 93.7 Hz, 3 J FF trans = 109.4 Hz, RhCCF cis to Rh), −140.4 (ddt, 3 J FF trans = 110.3 Hz, 3 J FF cis = 3 J PF = 36.9 Hz, 2 J RhF = 15.0 Hz, RhCFC). 31 P{ 1 H} NMR (81.0 MHz, C 6 D 6 ): δ 6.0 (dd, 1 J RhP = 140.8 Hz, 3 J PF = 39.9 Hz). Reaction <strong>of</strong> [Rh(η 5 -Cp*)(η 2 -C 2 H 4 )(PMe 3 )] with ICF(CF 3 )- CF 2 CF 3 . PMe 3 (0.07 mmol) was added to a solution <strong>of</strong> [Rh(η 5 - Cp*)(η 2 -C 2 H 4 ) 2 ] (21 mg, 0.071 mmol) in C 6 D 6 (0.5 mL) in an NMR tube. The tube was closed and heated at 120 °C until the conversion <strong>of</strong> the starting complex into [Rh(η 5 -Cp*)(η 2 -C 2 H 4 )(PMe 3 )] was complete according to the 1 H and 31 P{ 1 H} NMR spectra (24 h). Then, ICF(CF 3 )CF 2 CF 3 was added (12 μL, 0.073 mmol). A fast color change from yellow to dark red was observed. After 24 h at room temperature, a crystalline orange-red solid precipitated. After measuring NMR spectra, the solution was removed and the solid was washed with toluene (3 × 0.5 mL) and Et 2 O(3× 1 mL) and dried under vacuum. In the 19 F NMR spectrum <strong>of</strong> the solution, the main signals corresponded to trans- and cis-octafluoro-2-butene: 71 19 F NMR (188.3 MHz, C 6 D 6 ): δ (trans isomer) −69.1 (m, 6 F, CF 3 ), −159.8 (m, 2 F, CF); (cis isomer) −66.7 (m, 6 F, CF 3 ), −141.6 (m, 2 F, CF). Data <strong>of</strong> the solid: 1 H NMR (300.1 MHz, CD 2 Cl 2 ,21°C): δ 1296 Article 13.8 (very br s, 1 H, F n+1 H − n ), 1.94 (t, 4 J PH = 3.2 Hz, 15 H, C 5 Me 5 ), 1.76 (m, 9 H, PMe 3 ); (−90 °C) δ 16.2 (br t, 1 J FH = 121 Hz, HF − 2 ), 13.7 (br d, 1 J FH = 352 Hz, H 2 F − 3 ), 1.83 (br s, C 5 Me 5 ), 1.64 (br s, 9 H, PMe 3 ). 19 F NMR (282.4 MHz, CD 2 Cl 2 ,21°C): δ −128.2 (very br s, SiF 2− 6 ), −165.4 (very br s, F n+1 H − n ); (−90 °C) δ −128.5 (br s, SiF 2− 6 ), −146.6 (br t, 1 J FH = 131.5 Hz, [FHFHF] − ), −149.5 (br d, 1 J FH = 123.6 Hz, [FHF] − ), −174.3 (br dd, 1 J FH = 350.0 Hz, 2 J FF = 130.9 Hz, [FHFHF] − ). 31 P{ 1 H} NMR (81.0 MHz, CD 2 Cl 2 ): δ 1.2 (d, 1 J RhP = 131.9 Hz). (+)ESI-MS: m/z 517 ([Rh(η 5 -Cp*)I(PMe 3 ) 2 ] + ); exact mass calcd for C 16 H 33 IP 2 Rh 517.0152, found 517.0171, Δ =3.7ppm. [Rh(η 5 -Cp)( n C 4 F 9 )I(PMe 3 )] (5). A solution <strong>of</strong> [Rh(η 5 -Cp)(η 2 -C 2 H 4 )- (PMe 3 )] (290 mg, 1.07 mmol) in n-pentane (10 mL) was treated with I n C 4 F 9 (0.19 mL, 1.08 mmol). The mixture was stirred for 10 min. An orange solid precipitated, which was filtered, washed with n-pentane (2 × 10 mL), and dried under vacuum (302 mg, 48%). Mp: 196− 198 °C. Anal. Calcd for C 12 H 14 F 9 IPRh: C, 24.43; H, 2.39. Found: C, 24.31; H, 2.44. 1 H NMR (200.1 MHz, CDCl 3 ): δ 5.52 (d, 2 J RhH =1.5 Hz, 5 H, C 5 H 5 ), 1.81 (d, 2 J PH =11.4Hz,9H,PMe 3 ). 13 C{ 1 H} NMR (100.8 MHz, C 6 D 6 ): δ 135.3 (m, CF 2 ), 117.9 (qt, 1 J FC = 288.1 Hz, 2 J FC = 34.1 Hz, CF 3 ), 114.9−106.1 (two overlapped multiplets, 2 CF 2 ), 90.4 (s, C 5 H 5 ), 21.0 (d, 1 J PC = 35.6 Hz, PMe 3 ). 19 F NMR (188.3 MHz, CDCl 3 ): δ −54.8 (AB d, 2 J FF = 254.2 Hz, 1 F, C α F A ), −66.5 (AB d, 2 J FF = 257.0 Hz, 1 F, C α F B ), −81.7 (br s, 3 F, CF 3 ), −110.0 (AB d, 2 J FF = 281.9Hz,1F,C β F A ), −111.8 (AB d, 2 J FF = 279.6 Hz, 1 F, C β F B ), −125.7 (m, 2 F, C γ F 2 ). 31 P{ 1 H} NMR (81.0 MHz, C 6 D 6 ): δ 9.8 (dddd, 1 J RhP = 146.0 Hz, J PF = 21.9, 9.5, and 6.4 Hz). [Rh(η 5 -Cp*){CH 2 CH 2 CF(CF 3 ) 2 }(CNXy)(PPh 3 )](OTf) (6). AgOTf (36 mg, 0.14 mmol) was added to a solution <strong>of</strong> 3a′ (116 mg, 0.14 mmol) in THF (9 mL). The mixture was stirred for 2 h at room temperature and evaporated to dryness. The residue was stirred with CH 2 Cl 2 (9 mL), and the suspension was filtered. XyNC (19 mg, 0.14 mmol) was added to the resulting orange solution. After stirring for 5 h at room temperature, the resulting light orange solution was evaporated to dryness. The residue was washed with Et 2 O(3× 5 mL) and dried under vacuum to give 5 as a yellowish-brown solid (106 mg, 87%). Anal. Calcd for C 43 H 43 F 10 NO 3 PRhS: C, 52.82; H, 4.43; N, 1.43; S, 3.28. Found: C, 52.53; H, 4.50; N, 1.51; S, 3.24. IR (Nujol, cm −1 ): ν(CN) 2133. 1 H NMR (400.9 MHz, CD 2 Cl 2 ): δ 7.59 (m, 3 H, H4 <strong>of</strong> Ph), 7.51 (m, 6 H, H3 <strong>of</strong> Ph), 7.31 (m, 7 H, H2 <strong>of</strong> Ph and H4 <strong>of</strong> Xy), 7.17 (d, 3 J HH = 7.6 Hz, 2 H, H3 <strong>of</strong> Xy), 2.37−2.22 (m, 2 H, CH 2 CF), 2.18 (s, 6 H, Me <strong>of</strong> Xy), 1.80−1.57 (m, 2 H, RhCH 2 ), 1.67 (d, 4 J PH = 2.9 Hz, 15 H, C 5 Me 5 ). 13 C{ 1 H} NMR (100.8 MHz, CDCl 3 ): δ 151.7 (dd, 1 J RhC = 72.1 Hz, 2 J PC = 25.2 Hz, CN), 135.2 (s, C2 <strong>of</strong> Xy), 133.3 (d, 2 J PC = 9.7 Hz, C2 or C3 <strong>of</strong> Ph), 132.1 (s, C4 <strong>of</strong> Ph), 130.4 (s, C4 <strong>of</strong> Xy), 129.3 (br d, 2 J PC = 48.2 Hz, C1 <strong>of</strong> Ph), 129.3 (d, 2 J PC = 10.5 Hz, C3 or C2 <strong>of</strong> Ph), 128.8 (s, C3 <strong>of</strong> Xy), 126.5 (s, C− N), 120.81 (qd, 1 J FC = 286.6 Hz, 2 J FC = 28.2 Hz, CF 3 ), 120.74 (qd, 1 J FC = 286.7 Hz, 2 J FC = 28.1 Hz, CF 3 ), 104.9 (d, 1 J RhC = 2.1 Hz, 2 J PC = 3.9 Hz, C 5 Me 5 ), 91.0 (d <strong>of</strong> septuplets, 1 J FC = 201.7 Hz, 2 J FC = 31.0 Hz, CF), 35.3 (d, 2 J FC = 21.5 Hz, CH 2 CF), 18.8 (s, MeAr), 9.3 (s, C 5 Me 5 ), 4.8 (dd, 1 J RhC = 23.4 Hz, 2 J PC = 9.4 Hz, RhCH 2 ). 19 F NMR (188.3 MHz, CDCl 3 ): δ −75.4 (dq, 3 J FF = 4 J FF = 8.6 Hz, CF 3 CF), −76.9 (dq, 3 J FF = 4 J FF = 8.6 Hz, CF 3 CF), −79.0 (s, OTf), −184.9 (m, CF). 31 P{ 1 H} NMR (81.0 MHz, CDCl 3 ): δ 43.5 (d, 1 J RhP = 131.0 Hz). (+)ESI-MS: m/z 828 (M + ); exact mass calcd for C 42 H 43 F 7 NPRh 828.2071, found 828.2087, Δ = 1.9 ppm. [Rh(η 5 -Cp*){C(O)CH 2 CH 2 CF(CF 3 ) 2 }(CO)(PPh 3 )]OTf (7). AgOTf (32 mg, 0.12 mmol) was added to a solution <strong>of</strong> 3a′ (100 mg, 0.12 mmol) in THF (10 mL), and the mixture was stirred at room temperature for 2 h and evaporated to dryness. The residue was extracted with CH 2 Cl 2 (8 mL), and the extract was filtered. CO was bubbled through the extract for 3 min. Then, the reaction tube was closed and the solution was stirred for 48 h at room temperature. A small amount <strong>of</strong> black precipitate was removed by filtration, and the filtrate was evaporated to dryness to give a dark yellow solid, which was washed with n-pentane (3 × 3 mL) to give crude 7 (73 mg, 69%). Yellow, analytically pure crystals were obtained by liquid diffusion <strong>of</strong> n-pentane into a CH 2 Cl 2 solution. Mp: 144−146 °C. Anal. Calcd for C 36 H 34 F 10 O 5 PRhS: C, 47.91; H, 3.80; S, 3.55. Found: C, 48.23; H, 3.79; S, 3.35. IR (Nujol, cm −1 ): ν(CO) 2043 (CO), 1682 (CO). dx.doi.org/10.1021/om2009588 | Organometallics 2012, 31, 1287−1299
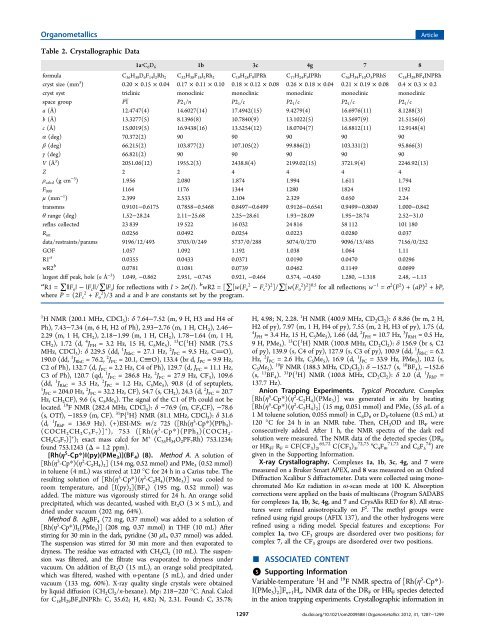
Organometallics Article Table 2. Crystallographic Data 1a·C 6 D 6 1b 3c 4g 7 8 formula C 36 H 38 D 6 F 14 I 2 Rh 2 C 32 H 38 F 18 I 2 Rh 2 C 19 H 28 F 9 IPRh C 17 H 24 F 9 IPRh C 36 H 34 F 10 O 5 PRhS C 18 H 29 BF 4 INPRh cryst size (mm 3 ) 0.20 × 0.15 × 0.04 0.17 × 0.11 × 0.10 0.18 × 0.12 × 0.08 0.26 × 0.18 × 0.04 0.21 × 0.19 × 0.08 0.4 × 0.3 × 0.2 cryst syst triclinic monoclinic monoclinic monoclinic monoclinic monoclinic space group P1̅ P2 1 /n P2 1 /c P2 1 /c P2 1 /c P2 1 /c a (Å) 12.4747(4) 14.6027(14) 17.4942(15) 9.4279(4) 16.6976(11) 8.1288(3) b (Å) 13.3277(5) 8.1396(8) 10.7840(9) 13.1022(5) 13.5697(9) 21.5156(6) c (Å) 15.0019(5) 16.9438(16) 13.5254(12) 18.0704(7) 16.8812(11) 12.9148(4) α (deg) 70.372(2) 90 90 90 90 90 β (deg) 66.215(2) 103.877(2) 107.105(2) 99.886(2) 103.331(2) 95.866(3) γ (deg) 66.821(2) 90 90 90 90 90 V (Å 3 ) 2051.06(12) 1955.2(3) 2438.8(4) 2199.02(15) 3721.9(4) 2246.92(13) Z 2 2 4 4 4 4 ρ calcd (g cm −3 ) 1.956 2.080 1.874 1.994 1.611 1.794 F 000 1164 1176 1344 1280 1824 1192 μ (mm −1 ) 2.399 2.533 2.104 2.329 0.650 2.24 transmns 0.9101−0.6175 0.7858−0.5468 0.8497−0.6499 0.9126−0.6541 0.9499−0.8049 1.000−0.842 θ range (deg) 1.52−28.24 2.11−25.68 2.25−28.61 1.93−28.09 1.95−28.74 2.52−31.0 reflns collected 23 839 19 522 16 032 24 816 58 112 101 180 R int 0.0256 0.0492 0.0254 0.0223 0.0280 0.037 data/restraints/params 9196/12/493 3703/0/249 5737/0/288 5074/0/270 9096/13/485 7156/0/252 GOF 1.057 1.092 1.192 1.038 1.064 1.11 R1 a 0.0355 0.0433 0.0371 0.0190 0.0470 0.0296 wR2 b 0.0781 0.1081 0.0739 0.0462 0.1149 0.0699 largest diff peak, hole (e Å −3 ) 1.049, −0.862 2.951, −0.745 0.921, −0.464 0.574, −0.450 1.280, −1.318 2.48, −1.13 a R1 = ∑||F o | − |F c ||/∑|F o | for reflections with I >2σ(I). b wR2 = [∑[w(F 2 o − F 2 c ) 2 ]/∑[w(F 2 o ) 2 ] 0.5 for all reflections; w −1 = σ 2 (F 2 )+(aP) 2 + bP, where P =(2F 2 c + F 2 o )/3 and a and b are constants set by the program. 1 H NMR (200.1 MHz, CDCl 3 ): δ 7.64−7.52 (m, 9 H, H3 and H4 <strong>of</strong> Ph), 7.43−7.34 (m, 6 H, H2 <strong>of</strong> Ph), 2.93−2.76 (m, 1 H, CH 2 ), 2.46− 2.29 (m, 1 H, CH 2 ), 2.18−1.99 (m, 1 H, CH 2 ), 1.78−1.64 (m, 1 H, CH 2 ), 1.72 (d, 4 J PH = 3.2 Hz, 15 H, C 5 Me 5 ). 13 C{ 1 H} NMR (75.5 MHz, CDCl 3 ): δ 229.5 (dd, 1 J RhC = 27.1 Hz, 2 J PC = 9.5 Hz, CO), 190.0 (dd, 1 J RhC = 76.2, 2 J PC = 20.1, CO), 133.4 (br d, J PC = 9.9 Hz, C2 <strong>of</strong> Ph), 132.7 (d, J PC = 2.2 Hz, C4 <strong>of</strong> Ph), 129.7 (d, J PC = 11.1 Hz, C3 <strong>of</strong> Ph), 120.7 (qd, 1 J FC = 286.8 Hz, 2 J FC = 27.9 Hz, CF 3 ), 109.6 (dd, 1 J RhC = 3.5 Hz, 2 J PC = 1.2 Hz, C 5 Me 5 ), 90.8 (d <strong>of</strong> septuplets, 1 J FC = 204.0 Hz, 2 J FC = 32.2 Hz, CF), 54.7 (s, CH 2 ), 24.3 (d, 2 J FC = 20.7 Hz, CH 2 CF), 9.6 (s, C 5 Me 5 ). The signal <strong>of</strong> the C1 <strong>of</strong> Ph could not be located. 19 F NMR (282.4 MHz, CDCl 3 ): δ −76.9 (m, CF 3 CF), −78.6 (s, OTf), −185.9 (m, CF). 31 P{ 1 H} NMR (81.1 MHz, CDCl 3 ): δ 31.6 (d, 1 J RhP = 136.9 Hz). (+)ESI-MS: m/z 725 ([Rh(η 5 -Cp*)(PPh 3 )- (COCH 2 CH 2 C 3 F 7 )] + ), 753 ([Rh(η 5 -Cp*)(PPh 3 )(COCH 2 - CH 2 C 3 F 7 )] + ); exact mass calcd for M + (C 35 H 34 O 2 PF 7 Rh) 753.1234; found 753.1243 (Δ = 1.2 ppm). [Rh(η 5 -Cp*)I(py)(PMe 3 )](BF 4 )(8). Method A. A solution <strong>of</strong> [Rh(η 5 -Cp*)(η 2 -C 2 H 4 ) 2 ] (154 mg, 0.52 mmol) and PMe 3 (0.52 mmol) in toluene (4 mL) was stirred at 120 °C for 24 h in a Carius tube. The resulting solution <strong>of</strong> [Rh(η 5 -Cp*)(η 2 -C 2 H 4 )(PMe 3 )] was cooled to room temperature, and [I(py) 2 ](BF 4 ) (195 mg, 0.52 mmol) was added. The mixture was vigorously stirred for 24 h. An orange solid precipitated, which was decanted, washed with Et 2 O(3× 5 mL), and dried under vacuum (202 mg, 64%). Method B. AgBF 4 (72 mg, 0.37 mmol) was added to a solution <strong>of</strong> [Rh(η 5 -Cp*)I 2 (PMe 3 )] (208 mg, 0.37 mmol) in THF (10 mL) After stirring for 30 min in the dark, pyridine (30 μL, 0.37 mmol) was added. Thesuspensionwasstirredfor30minmoreandthenevaporatedto dryness. The residue was extracted with CH 2 Cl 2 (10 mL). The suspension was filtered, and the filtrate was evaporated to dryness under vacuum. On addition <strong>of</strong> Et 2 O (15 mL), an orange solid precipitated, which was filtered, washed with n-pentane (5 mL), and dried under vacuum (133 mg, 60%). X-ray quality single crystals were obtained by liquid diffusion (CH 2 Cl 2 /n-hexane). Mp: 218−220 °C. Anal. Calcd for C 18 H 29 BF 4 INPRh: C, 35.62; H, 4.82; N, 2.31. Found: C, 35.78; 1297 H, 4.98; N, 2.28. 1 H NMR (400.9 MHz, CD 2 Cl 2 ): δ 8.86 (br m, 2 H, H2 <strong>of</strong> py), 7.97 (m, 1 H, H4 <strong>of</strong> py), 7.55 (m, 2 H, H3 <strong>of</strong> py), 1.75 (d, 4 J PH = 3.4 Hz, 15 H, C 5 Me 5 ), 1.66 (dd, 2 J PH = 10.7 Hz, 3 J RhH = 0.5 Hz, 9 H, PMe 3 ). 13 C{ 1 H} NMR (100.8 MHz, CD 2 Cl 2 ): δ 156.9 (br s, C2 <strong>of</strong> py), 139.9 (s, C4 <strong>of</strong> py), 127.9 (s, C3 <strong>of</strong> py), 100.9 (dd, 1 J RhC = 6.2 Hz, 2 J PC = 2.6 Hz, C 5 Me 5 ), 16.9 (d, 1 J PC = 33.9 Hz, PMe 3 ), 10.2 (s, C 5 Me 5 ). 19 F NMR (188.3 MHz, CD 2 Cl 2 ): δ −152.7 (s, 10 BF 4 ), −152.6 (s, 11 BF 4 ). 31 P{ 1 H} NMR (100.8 MHz, CD 2 Cl 2 ): δ 2.0 (d, 1 J RhP = 137.7 Hz). Anion Trapping Experiments. Typical Procedure. Complex [Rh(η 5 -Cp*)(η 2 -C 2 H 4 )(PMe 3 )] was generated in situ by heating [Rh(η 5 -Cp*)(η 2 -C 2 H 4 ) 2 ] (15 mg, 0.051 mmol) and PMe 3 (55 μL <strong>of</strong>a 1 M toluene solution, 0.055 mmol) in C 6 D 6 or D 8 -toluene (0.5 mL) at 120 °C for 24 h in an NMR tube. Then, CH 3 OD and IR F were consecutively added. After 1 h, the NMR spectra <strong>of</strong> the dark red solution were measured. The NMR data <strong>of</strong> the detected species (DR F or HR F ;R F = CF(CF 3 ) 2 , 65,72 C(CF 3 ) 3 , 72,73 n C 4 F 9 , 71,72 and C 6 F 74 5 ) are given in the Supporting Information. X-ray Crystallography. <strong>Complexes</strong> 1a, 1b, 3c, 4g, and 7 were measured on a Bruker Smart APEX, and 8 was measured on an Oxford Diffraction Xcalibur S diffractometer. Data were collected using monochromated Mo Kα radiation in ω-scan mode at 100 K. Absorption corrections were applied on the basis <strong>of</strong> multiscans (Program SADABS for complexes 1a, 1b, 3c, 4g, and 7 and CrysAlis RED for 8). All structures were refined anisotropically on F 2 . The methyl groups were refined using rigid groups (AFIX 137), and the other hydrogens were refined using a riding model. Special features and exceptions: For complex 1a, two CF 3 groups are disordered over two positions; for complex ■ 7, all the CF 3 groups are disordered over two positions. ASSOCIATED CONTENT *S Supporting Information Variable-temperature 1 Hand 19 F NMR spectra <strong>of</strong> [Rh(η 5 -Cp*)- I(PMe 3 ) 2 ]F n+1 H n . NMR data <strong>of</strong> the DR F or HR F species detected in the anion trapping experiments. Crystallographic information in dx.doi.org/10.1021/om2009588 | Organometallics 2012, 31, 1287−1299