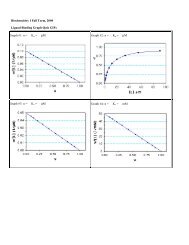
Practical Course on Multiple Sequence Alignment - CNB - Protein ...
Practical Course on Multiple Sequence Alignment - CNB - Protein ...
Practical Course on Multiple Sequence Alignment - CNB - Protein ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Choosing the right MSA software<br />
Given the multitude of choices, it can be difficult for a user of multiple<br />
alignment software to understand the situati<strong>on</strong>s in which a particular<br />
alignment tool is or is not appropriate. When aligning a small number (40%), most<br />
modern alignment programs will have no difficulty in returning a correct<br />
multiple sequence alignment, and no special c<strong>on</strong>siderati<strong>on</strong> is needed. When all<br />
of these c<strong>on</strong>diti<strong>on</strong>s do not hold, however, choosing the appropriate tools and<br />
c<strong>on</strong>figurati<strong>on</strong>, while keeping in mind the tradeoff between accuracy and<br />
computati<strong>on</strong>al cost, can be difficult. You can find a list of currently popular<br />
alignment software (see Table 2) and advice <strong>on</strong> tool selecti<strong>on</strong> (see Fig. 5).<br />
For more advanced discussi<strong>on</strong> <strong>on</strong> software selecti<strong>on</strong> see (Do & Katoh, 2008).<br />
Figure 5: Decisi<strong>on</strong> tree for choosing the right MSA program.<br />
<strong>Alignment</strong> visualizati<strong>on</strong> and editing<br />
Once an alignment has been generated, visualizati<strong>on</strong> tools allow manual<br />
identificati<strong>on</strong> of regi<strong>on</strong>s with reliably predicted homology; many of these tools<br />
also allow for interactive alignment editing.<br />
For alignments of sequences with low similarity, post-processing is extremely<br />
important as most regi<strong>on</strong>s in a low-identity alignment will not be reliably<br />
alignable.<br />
Visual inspecti<strong>on</strong> of an alignment and subsequent identificati<strong>on</strong> of potentially<br />
mis-aligned regi<strong>on</strong>s can be greatly helped using hints provided by software<br />
that highlight such regi<strong>on</strong>s. Typically, high c<strong>on</strong>fidence aligned regi<strong>on</strong>s can be<br />
identified by looking for groups of residues with str<strong>on</strong>gly c<strong>on</strong>served<br />
18