Extent of Absorption
Extent of Absorption
Extent of Absorption
- No tags were found...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
<strong>Extent</strong> <strong>of</strong> <strong>Absorption</strong><br />
The extent <strong>of</strong> absorption is important in determining the total<br />
body exposure or internal dose, and therefore is an important<br />
variable during chronic toxicity studies and/or chronic human<br />
exposure. The extent <strong>of</strong> absorption depends on the extent to<br />
which the chemical is transferred from the site <strong>of</strong><br />
administration into the local tissue, and the extent to which it<br />
is metabolized or broken down by local tissues prior to<br />
reaching the general circulation. An additional variable<br />
affecting the extent <strong>of</strong> absorption is the rate <strong>of</strong> removal from<br />
the site <strong>of</strong> administration by other processes compared with the<br />
rate <strong>of</strong> absorption<br />
١٨
Chemicals given via the gastrointestinal tract may be<br />
subject to a wide range <strong>of</strong> pH values and<br />
metabolizing enzymes in the gut lumen, gut wall, and<br />
liver before they reach the general circulation. The<br />
initial loss <strong>of</strong> chemical prior to it ever entering the<br />
blood is termed first-pass metabolism or pre-<br />
systemic metabolism; ; it may in some cases remove<br />
up to 100% <strong>of</strong> the administered dose so that none <strong>of</strong><br />
the parent chemical reaches the general circulation.<br />
The intestinal lumen contains a range <strong>of</strong> hydrolytic<br />
enzymes involved in the digestion <strong>of</strong> nutrients. The<br />
gut wall can perform similar hydrolytic reactions and<br />
contains enzymes that can oxidize many drugs<br />
١٩
٢٠
<strong>Absorption</strong> and Bioavailability<br />
Irrespective <strong>of</strong> the reason that is responsible for the incomplete<br />
absorption <strong>of</strong> the chemical as the parent compound, it is<br />
essential that there is a parameter which defines the extent <strong>of</strong><br />
transfer <strong>of</strong> the intact chemical from the site <strong>of</strong> administration<br />
into the general circulation. This parameter is the<br />
bioavailability, , which is simply the fraction <strong>of</strong> the dose<br />
administered that reaches the general circulation as the parent<br />
compound. (The term bioavailability is perhaps the most<br />
misused <strong>of</strong> all kinetic parameters and is sometimes used<br />
incorrectly in a general sense as the amount <strong>of</strong> drug available<br />
specifically to the site <strong>of</strong> toxicity.)<br />
٢١
Calculation <strong>of</strong> Bioavailability<br />
The fraction absorbed as the intact compound or bioavailability (F)) is<br />
determined by comparison with intravenous (i.v(<br />
i.v.) .) dosing (where F = 1 by<br />
definition). The bioavailability can be determined from the area under the<br />
plasma concentration–time time curve (AUC) <strong>of</strong> the parent compound , or the<br />
percentage dose excreted in urine as the parent compound, i.e. for an oral<br />
dose:<br />
٢٢
٢٣
2. Distribution<br />
Distribution is the reversible transfer <strong>of</strong> the<br />
chemical between the general circulation and<br />
the tissues. Irreversible processes such as<br />
excretion, metabolism, or covalent binding are<br />
part <strong>of</strong> elimination and do not contribute to<br />
distribution parameters. The important<br />
distribution parameters relate to the rate and<br />
extent <strong>of</strong> distribution.<br />
٢٤
Rate <strong>of</strong> Distribution<br />
The rate at which a chemical may enter or leave a tissue may be<br />
limited by two factors:<br />
(i) the ability <strong>of</strong> the compound to cross cell membranes and<br />
(ii) the blood flow to the tissues in which the chemical<br />
accumulates.<br />
The rate <strong>of</strong> distribution <strong>of</strong> highly water-soluble compounds may<br />
be slow due to their slow transfer from plasma into body<br />
tissues such as liver and muscle; water-soluble compounds do<br />
not accumulate in adipose tissue. In contrast, very lipid-soluble<br />
chemicals may rapidly cross cell membranes but the rate <strong>of</strong><br />
distribution may be slow because they accumulate in adipose<br />
tissue, and their overall distribution rate may be limited by<br />
blood flow to adipose tissue<br />
٢٥
The rate <strong>of</strong> distribution is indicated by the distribution<br />
rate constant, which is determined from the decrease<br />
in plasma concentrations in early time points after an<br />
intravenous dose. The rate constants refer to a mean<br />
rate <strong>of</strong> removal from the circulation and may not<br />
correlate with uptake into a specific tissue. Once an<br />
equilibrium has been reached between the general<br />
circulation and a tissue, any process which lowers the<br />
blood (plasma) concentration will cause a parallel<br />
decrease in the tissue concentration.<br />
٢٦
Factors affecting distribution<br />
1. Blood flow<br />
Drugs are readily distributed to highly perfused tissue<br />
like brain, liver, and kidneys<br />
2. Permeability limitations<br />
Many drugs do not readily enter the brain due to the<br />
blood brain barrier<br />
3. Protein binding<br />
Acidic drugs are bound to the most abundant plasma<br />
protein (albumin); while basic drugs bind to -1-<br />
acid glycoprotein.<br />
٢٧
4. Effect <strong>of</strong> pH<br />
The pH <strong>of</strong> the blood or tissue affect the<br />
ionization <strong>of</strong> the drug and thus its distribution<br />
5. Age<br />
In old people, Protein binding and body water<br />
will decrease, thus increasing the concentration<br />
<strong>of</strong> the drug per unit time<br />
6. Existence <strong>of</strong> storage sites:<br />
These include: Adipose tissue, plasma proteins,<br />
liver and kidneys, and bone<br />
٢٨
<strong>Extent</strong> <strong>of</strong> Distribution<br />
The extent <strong>of</strong> tissue distribution <strong>of</strong> a chemical depends<br />
on the relative affinity <strong>of</strong> the blood or plasma<br />
compared with the tissues. Highly water-soluble<br />
compounds that are unable to cross cell membranes<br />
readily are largely restricted to extracellular fluid<br />
(about 13 L per 70 kg body weight). Water-soluble<br />
compounds capable <strong>of</strong> crossing cell membranes (e.g.(<br />
caffeine, ethanol) are largely present in total body<br />
water (about 41 L per 70 kg body weight).<br />
٢٩
Lipid-soluble compounds frequently show extensive<br />
uptake into tissues and may be present in the lipids <strong>of</strong><br />
cell membranes, adipose tissue.<br />
The partitioning between circulating lipoproteins and<br />
tissue constituents is complex and may result in<br />
extremely low plasma concentrations. A factor which<br />
may further complicate the plasma/tissue partitioning<br />
is that some chemicals bind reversibly to circulating<br />
proteins such as albumin (for acid molecules) and<br />
acid glycoprotein (for basic molecules).<br />
٣٠
The extent and pattern <strong>of</strong> tissue distribution can<br />
be investigated by direct measurement <strong>of</strong><br />
tissue concentrations in animals. Tissue<br />
concentrations cannot be measured in human<br />
studies and, therefore, the extent <strong>of</strong> distribution<br />
in humans has to be determined based solely<br />
on the concentrations remaining in plasma or<br />
blood after distribution is complete.<br />
٣١
The parameter used to reflect the extent <strong>of</strong> distribution is the<br />
apparent volume <strong>of</strong> distribution (V),(<br />
which relates the total<br />
amount <strong>of</strong> the chemical in the body (Ab(<br />
Ab) ) to the circulating<br />
concentration (C)(<br />
) at any time after distribution is complete:<br />
The volumes <strong>of</strong> distribution <strong>of</strong> tubocurarine and caffeine are<br />
about 13 and 41 L per 70 kg because <strong>of</strong> their restricted<br />
distribution<br />
٣٢
Caffeine<br />
tubocurarine<br />
٣٣
when a chemical shows a more extensive reversible<br />
uptake into one or more tissues the plasma<br />
concentration will be lowered and the value <strong>of</strong> V will<br />
increase. For highly lipid-soluble chemicals, such as<br />
organochlorine pesticides, which accumulate in<br />
adipose tissue, the plasma concentration may be so<br />
low that the value <strong>of</strong> V may be many litres for each<br />
kilogram <strong>of</strong> body weight. This is not a real volume <strong>of</strong><br />
plasma and therefore V is called the apparent volume<br />
<strong>of</strong> distribution.<br />
٣٤
It is an important parameter because extensive<br />
reversible distribution into tissues, which will<br />
give a high value <strong>of</strong> V, , is associated with a low<br />
elimination rate and a long half-life life . It must be<br />
emphasized that the apparent volume <strong>of</strong><br />
distribution simply reflects the extent to which<br />
the chemical has moved out <strong>of</strong> the site <strong>of</strong><br />
measurement (the general circulation) into<br />
tissues, and it does not reflect uptake into any<br />
specific tissue(s).<br />
٣٥
Elimination<br />
The parameter most commonly used to describe<br />
the rate <strong>of</strong> elimination <strong>of</strong> a chemical is the<br />
half-life life . Most toxicokinetic processes are<br />
first-order reactions, i.e. the rate at which the<br />
process occurs is proportional to the amount <strong>of</strong><br />
chemical present. High rates (expressed as<br />
mass/time) occur at high concentrations and<br />
the rate decreases as the concentration<br />
decreases; in consequence the decrease is an<br />
exponential curve.<br />
٣٦
The usual way to analyze exponential changes is to use<br />
logarithmically transformed data which converts an<br />
exponential into a straight line. The slope <strong>of</strong> the line<br />
is the rate constant (k)(<br />
) for the process and the half-life<br />
life<br />
for the process is calculated as 0.693/k. . Rate<br />
constants and half-lives lives can be determined for<br />
absorption, distribution, and elimination processes.<br />
The clearance <strong>of</strong> a chemical is determined by the<br />
ability <strong>of</strong> the organs <strong>of</strong> elimination (e.g.(<br />
the liver,<br />
kidney, or lungs) to extract the chemical from the<br />
plasma or blood and permanently remove it by<br />
metabolism or excretion.<br />
٣٧
The mechanisms <strong>of</strong> elimination depend on the<br />
chemical characteristics <strong>of</strong> the compound:<br />
• volatile chemicals are exhaled,<br />
• water-soluble chemicals are eliminated in the<br />
urine and/or bile and<br />
• lipid-soluble chemicals are eliminated by<br />
metabolism to more water-soluble molecules,<br />
which are then eliminated in the urine and/or<br />
bile.<br />
٣٨
٣٩
If a chemical undergoes metabolic activation then<br />
toxicokinetic studies should measure both the parent<br />
chemical and the active metabolite. If the metabolite<br />
is so reactive that it does not leave the tissue in which<br />
it is produced (e.g.(<br />
alkylating metabolites <strong>of</strong> chemical<br />
carcinogens), then toxicokinetic studies should define<br />
the delivery <strong>of</strong> the parent chemical to the tissues, and<br />
the process <strong>of</strong> local activation should be regarded as<br />
part <strong>of</strong> tissue sensitivity (toxicodynamics(<br />
toxicodynamics) ) because it<br />
is not part <strong>of</strong> toxicokinetics, i.e. the movement <strong>of</strong> the<br />
chemical and/or metabolites around the body.<br />
٤٠
• Clearance = a ratio relating the rate <strong>of</strong><br />
elimination <strong>of</strong> a chemical from an appropriate<br />
reference fluid (usually plasma) to its<br />
concentration in the same reference fluid.<br />
Clearance has the units <strong>of</strong> flow rate in<br />
milliliters per minutes (mL(<br />
mL/min).<br />
• A clearance <strong>of</strong> 100 mL/minute <strong>of</strong> a chemical<br />
means that 100 mL <strong>of</strong> blood/plasma is<br />
completely cleared <strong>of</strong> the compound in each<br />
minute.<br />
٤١
The best measure <strong>of</strong> the ability <strong>of</strong> the organs <strong>of</strong><br />
elimination to remove the compound from the<br />
body is the clearance (CL(<br />
CL):<br />
Because the rate <strong>of</strong> elimination is proportional to the concentration,<br />
clearance is a constant for first-order processes and is independent <strong>of</strong><br />
dose. It can be regarded as the volume <strong>of</strong> plasma (or blood) cleared <strong>of</strong><br />
compound within a unit <strong>of</strong> time (e.g. mL/ min).<br />
٤٢
Renal clearance depends on the extent <strong>of</strong> protein<br />
binding, tubular secretion and passive reabsorption in<br />
the renal tubule; it can be measured directly from the<br />
concentrations present in plasma and urine:<br />
The total clearance or plasma clearance (which is the<br />
sum <strong>of</strong> all elimination processes, i.e. renal metabolic,<br />
etc.) ) is possibly the most important toxicokinetic<br />
parameter.<br />
٤٣
It is measured from the total amount <strong>of</strong> compound available for<br />
removal (i.e.(<br />
an intravenous dose) and the total area under the<br />
plasma concentration–time time curve (AUC) extrapolated to<br />
infinity.<br />
Plasma clearance reflects the overall ability <strong>of</strong> the body to remove<br />
permanently the chemical from the plasma. Plasma clearance is the<br />
parameter that is altered by factors such as enzyme induction, liver<br />
disease, kidney disease, inter-individual or inter-species differences in<br />
hepatic enzymes or in some cases organ blood flow.<br />
٤٤
Once the chemical is in the general circulation, the same volume<br />
<strong>of</strong> plasma will be cleared <strong>of</strong> chemical per minute (i.e.(<br />
the<br />
clearance value) applies irrespective <strong>of</strong> the route <strong>of</strong> delivery <strong>of</strong> o<br />
chemical into the circulation. However, the bioavailability (F)(<br />
will determine the proportion <strong>of</strong> the dose reaching the general<br />
circulation. Therefore, bioavailability has to be taken into<br />
account if clearance is calculated from data from a non-<br />
intravenous route (e.g.(<br />
oral):<br />
٤٥
Measurement <strong>of</strong> dose/AUC for an oral dose determines CL/F, , which contains<br />
two potentially independent variables – the amount <strong>of</strong> chemical delivered<br />
to the blood from the site <strong>of</strong> administration and the clearance <strong>of</strong> chemical<br />
present in the blood. The overall rate <strong>of</strong> elimination, as indicated by the<br />
terminal half-life life (t(<br />
), is dependent on two physiologically related and<br />
independent variables:<br />
where CL is the ability to extract and remove irreversibly the compound from f<br />
the general circulation, and V the extent to which the compound has left the<br />
general circulation in a reversible equilibrium with tissues.<br />
٤٦
Chemicals that are extremely lipid-soluble and<br />
are sequestered in adipose tissue are<br />
eliminated slowly. Lipid soluble<br />
organochlorine compounds, which are not<br />
substrates for P450 oxidation, due to the<br />
blocking <strong>of</strong> possible sites <strong>of</strong> oxidation by<br />
chloro-substituents<br />
substituents, , are eliminated extremely<br />
slowly: for example, the half-life life <strong>of</strong> 2,3,7,8-<br />
tetrachlorodibenzodioxin (TCDD) is about 8<br />
years in humans.<br />
٤٧
Summary <strong>of</strong> Toxicokinetic<br />
Parameters<br />
• Apparent volume <strong>of</strong> distribution (V(<br />
d )<br />
• A way to express the apparent space in the body that a chemical<br />
occupies<br />
• Expressed as a proportionality constant (in units <strong>of</strong> volume [e.g.,<br />
.,<br />
liters <strong>of</strong> blood] or volume/body weight)<br />
• Relates the total amount <strong>of</strong> chemical in the body to the<br />
concentration in plasma<br />
• For a 70-kg human,<br />
• Plasma volume ≈ 3 L (~0.045 L/kg body weight)<br />
• Total blood volume ≈ 5 L (~0.07 L/kg body weight)<br />
• Total extracellular fluid volume ≈ 12 L (~0.2 L/kg body weight)<br />
• Total body water ≈ 42 L (~0.6L/kg body weight)<br />
٤٨
• Example – If you know that 3 mg <strong>of</strong> chemical has been<br />
injected into the body, and the concentration <strong>of</strong> the<br />
chemical in the plasma is 1 mg/L, then you can calculate<br />
that the apparent V d to be 3 L. This suggests that the<br />
chemical is confined to the plasma space, and is not<br />
significantly absorbed into the tissues or into the<br />
particulate components (i.e., blood cells) in blood.<br />
3 mg ÷ 1.0 mg/L = 3.0 L<br />
٤٩
• Example – If you know that 3 mg <strong>of</strong> chemical has been<br />
injected and the concentration <strong>of</strong> the chemical in the<br />
plasma is 0.55 mg/L, then the apparent V d = 5.5 L. This<br />
suggests that the chemical may be distributed evenly<br />
between the plasma and the blood cells. More likely,<br />
the reality is that the chemical is somewhat absorbed<br />
into the tissues.<br />
3 mg ÷ 0.55 mg/L = 5.5 L<br />
٥٠
• Example – If you know that 3 mg <strong>of</strong> chemical has been<br />
injected and the concentration <strong>of</strong> the chemical in the<br />
plasma is 0.25 mg/L, then the apparent V d = 12 L. This<br />
suggests that the chemical may be distributed evenly<br />
within the total extracellular water. More likely, the<br />
reality is that the chemical is somewhat absorbed into<br />
the tissues, and perhaps somewhat metabolized.<br />
3 mg ÷ 0.25 mg/L = 12 L<br />
• It is possible to have a V d greater than the total body<br />
water<br />
• A high V d is an indication that one or both <strong>of</strong> two things<br />
has occurred,<br />
• The chemical has been absorbed from the blood and<br />
concentrated in the tissues.<br />
• The chemical has been metabolized.<br />
٥١
• Clearance (Cl(<br />
Cl)<br />
• A ratio relating the rate <strong>of</strong> elimination <strong>of</strong> a chemical<br />
from an appropriate reference fluid (usually plasma) to<br />
its concentration in the same reference fluid.<br />
• Has the units <strong>of</strong> flow rate in volume cleared per unit<br />
time (e.g., mL/min)<br />
• A clearance <strong>of</strong> 100 mL/minute <strong>of</strong> a chemical means that<br />
100 mL <strong>of</strong> blood/plasma is completely cleared <strong>of</strong> the<br />
compound in each minute .<br />
٥٢
• Zero- versus first-order kinetics<br />
• For zero order kinetics, the rate <strong>of</strong> elimination <strong>of</strong> a compound is a<br />
constant, and is independent <strong>of</strong> the concentration <strong>of</strong> the chemical l in<br />
the blood. For example, the average human body is able to<br />
eliminate ~10-15<br />
15 mL <strong>of</strong> ethanol per hour, regardless <strong>of</strong> the amount<br />
<strong>of</strong> ethanol consumed. This may be higher or lower depending on<br />
the factors previously discussed (e.g., induction <strong>of</strong> alcohol<br />
dehydrogenase by repeated alcohol exposure).<br />
• For first-order kinetics, the rate <strong>of</strong> elimination <strong>of</strong> a compound is<br />
dependent on the concentration <strong>of</strong> the chemical in the blood. The<br />
higher the concentration, the more rapidly the chemical is<br />
eliminated, unless the elimination mechanisms have been saturated.<br />
At that point, the kinetics become zero-order. order. This is known as<br />
saturation kinetics.<br />
٥٣
• Half-life life (t 1/2<br />
1/2 )<br />
• The time required for the concentration <strong>of</strong> a chemical in<br />
the plasma to decrease by 50%.<br />
• This is a constant for all but zero-order order kinetics. For<br />
example, for a compound eliminated by first-order<br />
kinetics, if the concentration at time 0 is 4 mg/L, and the<br />
t 1/2 is 6 hours, then at the end <strong>of</strong> 6 hours, the plasma<br />
concentration will be (0.5 x 4 mg/mL<br />
mL =) 2 mg/L, and at<br />
the end <strong>of</strong> the next 6 hours, the concentration will be<br />
(0.5 x 2 mg/mL<br />
mL =) 1 mg/L, and so on.<br />
٥٤
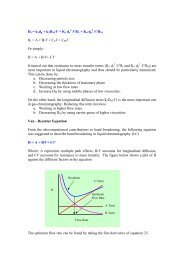
• Area under the Curve (AUC)<br />
• A measure <strong>of</strong> the total amount <strong>of</strong> chemical present in<br />
the body over a defined period. This is defined as the<br />
area under the concentration vs time curve for the<br />
defined period.<br />
• Example: The shaded area in the figure below<br />
represents the AUC(0-6hr). You can express the AUC<br />
for any given time period. It is <strong>of</strong>ten expressed as the<br />
AUC(0-∞).<br />
•<br />
Concentration<br />
in<br />
plasma<br />
0<br />
Time (hrs)<br />
6<br />
٥٥
• Classical toxicokinetics<br />
• One compartment model<br />
• The elimination <strong>of</strong> a chemical is said to follow a one-<br />
compartment model when the elimination phase <strong>of</strong> the<br />
log concentration vs time curve is a straight line.<br />
Conceptually, this is as though the chemical were<br />
evenly distributed throughout a single body<br />
compartment (e.g., total body water), and is eliminated<br />
at a constant rate over time. That is, a constant<br />
percentage <strong>of</strong> the chemical present is eliminated over<br />
any given time period.<br />
٥٦
• The rate at which a chemical is eliminated at any time is directly<br />
proportional to the amount <strong>of</strong> that chemical in the body at that time.<br />
• A one-compartment model indicates that no one tissue has a high affinity<br />
for the chemical. Changes in the plasma reflect changes in the tissue.<br />
Plasma<br />
Concentratio<br />
n<br />
Log <strong>of</strong><br />
Plasma<br />
Concentratio<br />
n<br />
Time<br />
Time<br />
٥٧
• Two compartment model<br />
• The elimination <strong>of</strong> a chemical is said to follow a two-<br />
compartment model when the elimination phase <strong>of</strong> the<br />
log concentration vs time curve is not a straight line.<br />
Conceptually, this is as though the chemical were<br />
unevenly distributed throughout two body<br />
compartments (e.g., highly concentrated in tissues and a<br />
lower concentration in the plasma). The net elimination<br />
is a sum <strong>of</strong> the rates <strong>of</strong> elimination from the various<br />
compartments.<br />
• The appearance <strong>of</strong> a two-compartment curve suggests<br />
that the chemical is distributed out <strong>of</strong> the blood into<br />
tissues.<br />
٥٨
Alpha distribution<br />
Log <strong>of</strong> Plasma<br />
Concentration<br />
Beta distribution<br />
Time<br />
٥٩
Chronic Administration<br />
The kinetic concepts and parameters <strong>of</strong> a single dose (as<br />
discussed above) apply to chronic administration, but<br />
the exposure has to allow for the fact that not all <strong>of</strong><br />
the previous dose(s) ) may have been eliminated when<br />
the subsequent dose is given. Therefore, there may be<br />
an increase in plasma concentration (and body load)<br />
until an equilibrium is reached in which the rate <strong>of</strong><br />
elimination balances out the rate <strong>of</strong> input, in other<br />
words, the daily dose is eliminated each day.<br />
٦٠
٦١
٦٢
The equations above assume that CL is not<br />
altered by repeated exposure; the assumption is<br />
not correct if the chemical induces or inhibits<br />
its own elimination because clearance would<br />
be increased or decreased, respectively, after<br />
the period <strong>of</strong> chronic intake.<br />
٦٣
Because the plasma and therefore tissue concentrations<br />
increase during chronic intake until an equilibrium is<br />
reached , the amount in the body (Ab(<br />
Ab) ) will also<br />
increase to reach a steady state. The time taken to<br />
reach steady state is 4–54<br />
5 times the elimination half-<br />
life and, therefore, the true duration <strong>of</strong> steady-state<br />
state<br />
exposure in a toxicity study is the study duration<br />
minus 4–54<br />
5 half-lives lives <strong>of</strong> the chemical (needed to reach<br />
the steady state). This is particularly important for<br />
chemicals that have a very long half-life; life; for example<br />
in rodents the steady-state state body load <strong>of</strong> TCDD, which<br />
has a half-life life in rats <strong>of</strong> about 1 month, will not be<br />
reached until after about 4–54<br />
5 months <strong>of</strong> continuous<br />
treatment.<br />
٦٤
Saturation Kinetics<br />
All the parameters described above relate to first-order<br />
processes and, therefore, are independent <strong>of</strong> dose at<br />
low doses. However, at high doses and/or during<br />
chronic studies it is possible to overload or saturate<br />
compound–protein protein interactions. Under such<br />
circumstances any increase in the concentration <strong>of</strong> the<br />
compound cannot give a proportional (first-order)<br />
increase in the rate <strong>of</strong> the process. When a process is<br />
saturated the rate is at the maximum possible and is<br />
essentially independent <strong>of</strong> concentration.<br />
٦٥
In simple mathematical terms this means that the reaction<br />
changes from first to zero order. This is best described by<br />
Michaelis–Menten<br />
Menten kinetics, i.e.<br />
٦٦
3. Metabolism<br />
• Many xenobiotics undergo chemical transformation<br />
(biotransformation; metabolism) when introduced<br />
into biologic systems like the human body.<br />
• Biotransformation is <strong>of</strong>ten mediated by enzymes<br />
• End result <strong>of</strong> biotransformation is either alteration <strong>of</strong><br />
the parent molecule, or conjugation <strong>of</strong> the parent<br />
molecule (or its metabolites) with endogenous<br />
substances in the body.<br />
• Enzymes involved in biotransformation can act on<br />
either endogenous or xenobiotic compounds,<br />
especially if the xenobiotics are structurally similar to<br />
endogenous compounds<br />
٦٧
• Example: Cholinesterase is an enzyme that normally metabolizes<br />
acetylcholine, a neurotransmitter found in the synapse.<br />
Cholinesterase can also metabolize the local anesthetic agent<br />
procaine and the muscle-paralyzing agent succinylcholine. . If a<br />
person has high levels <strong>of</strong> cholinesterase activity, the effectiveness<br />
<strong>of</strong> these drugs can be compromised. If a person is exposed to<br />
anything that inhibits cholinesterase activity, the pharmacologic<br />
effects <strong>of</strong> the drugs can be enhanced, resulting in toxicity.<br />
• Example: Monoamine oxidase (MAO) is an enzyme that normally<br />
metabolizes biologic amines like epinephrine. MAO can also<br />
oxidize a variety <strong>of</strong> drugs. If a person is taking a drug that inhibits i<br />
MAO activity (like many blood pressure medications), it can be<br />
dangerous for that person to take other drugs that can be<br />
metabolized by MAO.<br />
٦٨
• The products <strong>of</strong> biotransformation can be either less toxic<br />
than, more toxic than, or about as toxic as the parent<br />
molecules.<br />
• Enzymes involved in biotransformation are sometimes called<br />
“drug metabolizing enzymes”. . Although strictly speaking this<br />
is a misnomer because many <strong>of</strong> the substrates are not drugs,<br />
the term is still commonly used.<br />
• Location <strong>of</strong> metabolic enzymes<br />
• Species – found in virtually every species, although the type and<br />
amount vary tremendously.<br />
• Organs – present in many tissues. Many enzymes are particularly<br />
abundant in the liver.<br />
• Subcellular – many <strong>of</strong> the drug-metabolizing enzymes are located in<br />
the smooth endoplasmic reticulum (SER). These are called<br />
“microsomal” enzymes.<br />
٦٩
• Types <strong>of</strong> Biotransformation Reactions<br />
• Basically, two types <strong>of</strong> reactions, nonsynthetic (Phase I) and<br />
synthetic (Phase II)<br />
• Phase I reactions<br />
• Involve modification <strong>of</strong> the basic structure <strong>of</strong> the substrate<br />
• Do not involve covalent binding <strong>of</strong> the substrate to an endogenous<br />
compound<br />
• Examples include hydrolysis, oxidation, and reduction reactions<br />
• Phase I enzymes are <strong>of</strong>ten membrane-bound bound (e.g.,<br />
microsomal). This is because they generally act on more lipid-<br />
soluble (nonpolar(<br />
nonpolar) ) substrates, and their purpose is to make the<br />
compounds MORE POLAR and therefore, MORE EASILY<br />
EXCRETABLE by the kidney and biliary tract. Think <strong>of</strong> the<br />
active transport mechanisms in the kidney.<br />
٧٠
• Oxidation<br />
• Uses molecular oxygen (O2). One atom <strong>of</strong> oxygen is<br />
combined with hydrogen to form water, and the other<br />
atom <strong>of</strong> oxygen is introduced into the substrate<br />
molecule.<br />
• Involves several enzymatic steps.<br />
• The oxidative system is <strong>of</strong>ten known as the “mixed<br />
function oxidase” system”. . These enzymes are some <strong>of</strong><br />
the most thoroughly researched enzymes in biological<br />
systems.<br />
• One subfamily <strong>of</strong> the mixed function oxidase system is the<br />
group <strong>of</strong> enzymes known as Cytochrome P-450<br />
enzymes.<br />
They are so called because <strong>of</strong> their absorbance characteristics<br />
at wavelengths <strong>of</strong> 448-450 450 nm.<br />
٧١
• Anything that affects the activity <strong>of</strong> any one <strong>of</strong> the steps can affect a<br />
the way the body reacts to a given drug or other xenobiotic.<br />
• Examples <strong>of</strong> the various types <strong>of</strong> oxidation reactions are in the<br />
textbook,<br />
• Deamination – replacement <strong>of</strong> an amine group (NH 2<br />
) with an oxygen<br />
(O) atom<br />
• N-, , O-, O , or S-DealkylationS<br />
– replacement <strong>of</strong> an alkyl group (e.g., CH 3<br />
)<br />
with a hydrogen atom. Typically, the alkyl group in the parent<br />
molecule is bonded to a N, O, or S atom.<br />
• Aliphatic or aromatic hydroxylation – addition <strong>of</strong> a hydroxyl group<br />
(OH) to a molecule<br />
• N-oxidation<br />
– replacement <strong>of</strong> a hydrogen atom on an amine with an<br />
oxygen<br />
• S-oxidation<br />
– addition <strong>of</strong> an oxygen atom to a sulfur atom<br />
• Conversion <strong>of</strong> a hydroxyl group (alcohol) to a carboxyl group (acid)<br />
٧٢
• Reduction<br />
• Azo reduction – reduction <strong>of</strong> an azo bond (N=N) to two<br />
amines (NH 2 )<br />
• Nitro reduction – reduction <strong>of</strong> a nitro group (NO 2 ) to an<br />
amine<br />
• Hydrolysis<br />
• Addition <strong>of</strong> water (H 2 O) to an ester bond (C-O-O-C) C) to<br />
form an alcohol (C-OH) and a carboxylic acid (COOH)<br />
R-C-O-O-C-R R + H-O-H H = R-C-OH + R-COOHR<br />
٧٣
• Phase II reactions<br />
• Involve addition <strong>of</strong> a c<strong>of</strong>actor to a substrate to form a new<br />
product. Therefore, the rate <strong>of</strong> these reactions can be<br />
limited by the availability <strong>of</strong> the c<strong>of</strong>actor.<br />
• Phase II enzymes may be either microsomal or cytosolic.<br />
This is because the primary purpose <strong>of</strong> the Phase II<br />
reactions is not so much to increase the polarity <strong>of</strong> the<br />
parent compound (although that is part <strong>of</strong> what they<br />
accomplish). The primary purpose is to increase the<br />
molecular weight <strong>of</strong> the parent compound to make it a<br />
better substrate for active transport mechanisms in the<br />
biliary tract.<br />
• Various factors can affect the availability <strong>of</strong> c<strong>of</strong>actors. For<br />
example, fasting markedly reduces the amount <strong>of</strong><br />
glutathione available in the liver.<br />
٧٤
• Sulfation<br />
• Replacement <strong>of</strong> a hydrogen atom (H) with a sulfonate (SO3)<br />
• Uses the enzyme sulfotransferase<br />
• Uses the c<strong>of</strong>actor called PAPS (phosphoadenosine(<br />
phosphosulfate)<br />
• Produces a highly water-soluble sulfuric acid ester<br />
• Glucuronidation<br />
• Replacement <strong>of</strong> a hydrogen atom with a glucuronic acid<br />
• Uses the enzyme UDP-glucuronosyl<br />
transferase (UDP-GT)<br />
• Uses the c<strong>of</strong>actor called UDPGA (uridine(<br />
diphosphate glucuronic<br />
acid)<br />
• One <strong>of</strong> the major Phase II enzymatic pathways<br />
٧٥
• Acetylation<br />
• Replacement <strong>of</strong> a hydrogen atom with an acetyl group<br />
• Uses the enzyme acetyltransferase<br />
• Uses the c<strong>of</strong>actor called acetyl CoA (acetyl coenzyme A)<br />
• Sometimes results in a less water-soluble product<br />
• Remember the discussion <strong>of</strong> slow versus fast acetylators.<br />
• Methylation<br />
• Replacement <strong>of</strong> a hydrogen atom with a methyl group<br />
• Uses the enzyme methyltransferase<br />
• Uses the c<strong>of</strong>actor called SAM (S-adenosyl<br />
methionine)<br />
• Common but relatively minor pathway<br />
٧٦
• Glutathione conjugation<br />
• Adds a glutathione molecule to the parent compound, either by<br />
direct addition or by replacement <strong>of</strong> an electrophilic substituent<br />
(e.g., a halogen atom)<br />
• Uses the enzyme glutathione transferase (GST)<br />
• Uses the c<strong>of</strong>actor called glutathione (a tripeptide made up <strong>of</strong><br />
glycine, cysteine, , and glutamic acid<br />
• One <strong>of</strong> the major Phase II enzymatic pathways<br />
• Amino acid conjugation<br />
• Adds an amino acid to the parent compound.<br />
• Mercapturic acid formation<br />
• Formed by cleavage <strong>of</strong> the glycine and glutamic acid substituents<br />
from a glutathione conjugate, followed by N-acetylationN<br />
<strong>of</strong> the<br />
resulting product<br />
٧٧
• Significance <strong>of</strong> Biotransformation Reactions<br />
in Toxicology<br />
• Biotransformation is a major part <strong>of</strong> the<br />
pathway for elimination <strong>of</strong> many xenobiotic<br />
compounds.<br />
• Biotransformation can result in either a<br />
decrease or an increase (or no change) in<br />
toxicity.<br />
• Biotransformation can result in the formation<br />
<strong>of</strong> reactive metabolites.<br />
٧٨
• Good example – metabolism <strong>of</strong> acetaminophen<br />
• Acetaminophen is ordinarily metabolized in the liver by sulfation and<br />
glucuronidation to form non-toxic conjugates<br />
• These are low capacity pathways, in that the c<strong>of</strong>actors are available able in<br />
only limited concentrations, so these are rate-limiting.<br />
• As long as the amount <strong>of</strong> acetaminophen in the liver is relatively y low,<br />
the Phase II pathways can handle the compound, and there is no<br />
toxicity.<br />
• If the concentration <strong>of</strong> acetaminophen becomes high enough to<br />
overwhelm the capacity <strong>of</strong> the Phase II pathways, an alternate<br />
metabolic pathway, involving Phase I enzymes, becomes active.<br />
• The product <strong>of</strong> the Phase I reaction is a highly reactive quinoneimine,<br />
which can form adducts with (bind covalently to) cellular<br />
macromolecules, especially proteins.<br />
• The binding <strong>of</strong> the reactive intermediate to cellular macromolecules<br />
les<br />
destroys the activity <strong>of</strong> those molecules, and can lead to compromised<br />
mised<br />
cell function and, ultimately, cell death.<br />
٧٩
• Another good example – metabolism <strong>of</strong> carbon<br />
tetrachloride<br />
• Carbon tetrachloride is metabolized by the<br />
cytochrome P-450 system in the liver by<br />
abstraction <strong>of</strong> one <strong>of</strong> the four chlorine atoms.<br />
• This results in formation <strong>of</strong> a highly reactive<br />
trichloromethane radical, which initiates a cascade<br />
<strong>of</strong> lipid peroxidation by removing a hydrogen atom<br />
from membrane phospholipids.<br />
• Damage to the cell membrane causes loss <strong>of</strong><br />
osmotic integrity, cell swelling and death.<br />
٨٠
• The activity <strong>of</strong> drug metabolizing enzymes is<br />
dependent on numerous factors<br />
• Species<br />
• Age (activity is generally lower in very young and aged<br />
animals)<br />
• Sex (activity is generally higher in males than in females)<br />
• Genetics (remember slow versus fast acetylators)<br />
• Organ (activity <strong>of</strong> many enzymes is highest in the liver)<br />
• General health status (e.g., hepatic injury decreases<br />
metabolic activity in the liver)<br />
• Diet (remember how fasting decreases the amount <strong>of</strong><br />
glutathione available for GST)<br />
• Previous exposure to other compounds<br />
٨١
• Induction – an increase in the activity <strong>of</strong> one or more<br />
enzymes as a result <strong>of</strong> previous exposure <strong>of</strong> the<br />
organism to compounds that serve as substrates for<br />
the enzyme(s)<br />
• Classic example <strong>of</strong> an inducer is phenobarbital, , which<br />
induces the activity <strong>of</strong> cytochrome P-450 enzymes<br />
• Induction may involve either increases in the synthesis <strong>of</strong><br />
enzymatic protein, or increases in activation <strong>of</strong><br />
proenzymes.<br />
• Induction may result in increases in the amount <strong>of</strong> cellular<br />
protein, hypertrophy (increases in size) <strong>of</strong> the cells, and<br />
increases in weight <strong>of</strong> the affected organs (especially liver).<br />
• One effect <strong>of</strong> induction <strong>of</strong> microsomal enzymes is an<br />
increase in the amount <strong>of</strong> smooth endoplasmic reticulum in<br />
a cell. This can be seen microscopically.<br />
٨٢
• Induction is usually temporary, and enzyme activity levels<br />
return to normal after several weeks.<br />
• Induction can result in tolerance to drugs, if the metabolism<br />
<strong>of</strong> the drugs results in a product with lower (or no)<br />
pharmacologic activity. This is why, for example, patients<br />
can develop tolerance to Phenobarbital anesthesia after<br />
repeated administration.<br />
• Induction may result in increases or decreases in toxicity,<br />
depending on whether the metabolite is more or less toxic<br />
than the parent compound. This is why, for example,<br />
alcoholics are more susceptible to acetaminophen toxicity,<br />
since alcohol induces the enzyme that is responsible for<br />
production <strong>of</strong> the reactive metabolite from acetaminophen.<br />
٨٣
• Inhibition – a decrease in the activity <strong>of</strong> one or more<br />
enzymes<br />
• Classic example <strong>of</strong> an inhibitor is SKF-525A, which<br />
inhibits microsomal enzymes<br />
• Inhibition may be either competitive or non-competitive.<br />
• Competitive inhibition<br />
• Occurs when an inhibitor binds to the same active site on the<br />
enzyme as the substrate. The higher the concentration <strong>of</strong> the<br />
inhibitor, the less likely it is that the substrate molecule will l be able<br />
to find and bind to an available enzyme molecule.<br />
• Reversible, since the binding <strong>of</strong> the inhibitor to the active site e is not<br />
covalent<br />
• Example: Omeprazole and diazepam are both metabolized by<br />
cytochrome P-450 2C19 (CYP2C19). Co-administration <strong>of</strong> these<br />
two drugs results in prolonged plasma half-life life for diazepam.<br />
٨٤
• Non-competitive inhibition<br />
• May occur when an inhibitor binds to the same active site on the enzyme<br />
as the substrate, but binds so tightly that it is effectively not t released. Thus,<br />
the binding site is permanently blocked.<br />
• May also occur when an inhibitor binds tightly (sometimes covalently) to a<br />
different site on the enzyme than the active site. This can result in<br />
conformational or affinity changes that effectively inactive the enzyme.<br />
• Non-competitive inhibition is generally not reversible. Therefore,<br />
recovery takes much longer because it requires the synthesis <strong>of</strong> new<br />
enzyme.<br />
• A special subset <strong>of</strong> non-competitive inhibition is “suicide inhibition”, , in<br />
which a compound binds to the active site <strong>of</strong> an enzyme and is<br />
metabolized, but the product then binds irreversibly to the active site. An<br />
example <strong>of</strong> this is the binding <strong>of</strong> organophosphate insecticides to the<br />
enzyme acetylcholinesterase (AchE). This results in a prolonged inhibition<br />
<strong>of</strong> AchE activity.<br />
• Inhibition may result in increases or decreases in toxicity, depending ending on<br />
whether the metabolite is more or less toxic than the parent compound.<br />
٨٥
Factors affecting metabolism<br />
1. Age<br />
The metabolizing enzymes in neonates are not fully<br />
developed, therefore those cannot efficiently<br />
metabolize drugs. Also in the elderly, enzymatic<br />
systems may not function well leading to same<br />
conclusion.<br />
2. Sex<br />
Males who are deficient in glucose -6-phosphate<br />
dehydrogenase are more prone to hemolysis when<br />
subjected to some drugs like sulfonamides<br />
٨٦
3. Pharmacogenetic factors<br />
Some individuals may be deficient in some enzymes, regardless<br />
<strong>of</strong> sex<br />
4. Pregnancy<br />
Hepatic metabolism <strong>of</strong> drugs is decreased in pregnancy<br />
5. Nutritional status/ liver dysfunction<br />
Malnutrition can cause a decreased level <strong>of</strong> some enzyme system<br />
and liver dysfunction can lead to decreased metabolism<br />
6. Bioactivation<br />
Some drugs may be transformed to more toxic metabolites<br />
7. Enzyme induction / inhibition<br />
A result <strong>of</strong> this is either an increase in the metabolism or a<br />
decrease in the drug metabolism<br />
8. Changes in the kinetic mechanism: depending on whether the<br />
concentration <strong>of</strong> drug is in the therapeutic or overdose range<br />
٨٧
4. Excretion<br />
• Excretion<br />
• The removal <strong>of</strong> materials from the body<br />
• Routes <strong>of</strong> excretion<br />
• Bile – through the liver<br />
• Urine – through the kidneys<br />
• Feces – through the intestines<br />
• Expired air – through the lungs<br />
• Sweat – through the skin<br />
• Saliva – through the mouth<br />
• Milk<br />
• Hair<br />
٨٨
Factors affecting excretion<br />
1. Age<br />
Many <strong>of</strong> the functions <strong>of</strong> the kidney are not well developed in<br />
neonates, toxic drugs will be slowly excreted. In the elderly,<br />
lower renal plasma flow will also decrease excretion<br />
2. Disease states<br />
Renal and gastrointestinal diseases can considerably slow the<br />
excretion<br />
3. Recirculation<br />
Some drugs that are excreted as their more water soluble<br />
metabolites may be back transformed to the parent<br />
compound by some bacteria<br />
4. Ion trapping<br />
Weakly acidic drugs trapped in the tubules are excreted at higher<br />
urinary pH as the ion form, thus preventing reabsorption.<br />
٨٩