Tema 12 - Medic.ula.ve
Tema 12 - Medic.ula.ve
Tema 12 - Medic.ula.ve
- No tags were found...

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
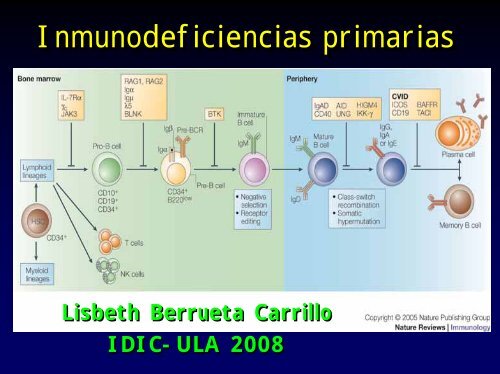
Inmunodeficiencias primariasLisbeth Berrueta CarrilloIDIC-ULA 2008
Abbas A et et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007• Primarias:Inmunodeficiencias– Errores en los mecanismos de defensa específicos oinespecíficos, derivados de alteraciones genéticas que conducena :• Deficiencia funcional de algunos de los elementos de la RI• Imposibilidad para el reconocimiento Ag• Alteración de la interconexión del SI• Mas de 200 enfermedades por inmunodeficiencias primarias• Secundarias:– La causa mas común de inmunodeficiencia:• Malnutrición, post-transfusión, infecciones crónicas• Secundaria a infecciones virales: HIV, sarampión,• Iatrogénica: tratamiento anti- cáncer, esteroides. esplenectomía
Inmunodeficiencias:Características generales• Predisposición a infecciones• Predisposición a desarrollar cáncer• Predisposición a desarrollar enfermedadesautoinmunes• Pueden resultar de defectos en lamaduración y activación de linfocitos o dedefectos en mecanismos efectores innatoso adquiridos
Inmunodeficiencias primarias:¿cuáles son las manifestaciones?• Manifestaciones altamente sugestivas:– Infección crónica (sinopulmonar)– Infección recurrente– Agentes infecciosos inusuales• Manifestaciones moderadamentesugestivas:– Diarrea crónica– Retardo del crecimiento y desarrollo– Hepato-esplenomegalia– Abscesos recurrentes
Inmunodeficiencias primarias:EVALUACIÓN CLÍNICA¿Cuales son los gérmenes mas frecuentes?• Historia clínica– Antecedentes personales:• Crecimiento y desarrollo• Respuesta a las inmunizaciones• Historia de infecciones• Fenómenos autoinmunes– Antecedentes familiares• Historia de infecciones• Fallecimiento a temprana edad
Inmunodeficiencias primarias:Frecuencia– Humorales 50%–Cel<strong>ula</strong>res 10%–Combinadas 18%– Cél<strong>ula</strong>s fagocíticas 20%– Complemento 2%
Abbas A et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007Clasificación de lasinmunodeficiencias• Por defectos en la maduración delinfocitos• Por defectos en la activación yfunción de linfocitos• Asociadas con otras enfermedadeshereditarias• Defectos en la inmunidad innata• Defectos en Inmunorreg<strong>ula</strong>ción
Inmunodeficiencias por defectosen la maduración de linfocitos• SCID (inmunodeficiencia combinada se<strong>ve</strong>ra)– SCID por mutación en cadena γc– Por deficiencia de ADA– Mutaciones en RAG1 y 2 Artemis, CD45– Disgenesia Retic<strong>ula</strong>r (defecto a ni<strong>ve</strong>l de Stemcell)• Defectos en la maduración de B– Aganmaglobulinemia ligada al X• Defectos en la maduración de T–Sindromede Di-George
Inmunodeficiencias primarias:Inmunodeficiencia combinada se<strong>ve</strong>ra(SCID)DisgenesiaRetic<strong>ula</strong>rIL-7IL-7Abbas A et et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007Candotti F et al JCI 2002; 109:<strong>12</strong>61
Ligada alcromosoma X:Mutación de lacadena gammacomún50% de SCIDRecesivasAfecta T y NK,
SCID secundaria a deficienciade Adenosindeaminasa (ADA)• Autosómica recesiva (25% del resto deSCID)• Acum<strong>ula</strong>ción de deoxiadenosina (inhibiciónde la síntesis del ADN)• Afecta el desarrollo de T y B, alrededor deun año de edad• Linfocitos T no proliferan en respuesta alantígeno– Deficiencia de purin nucleosido fosforilasa(PNP)
WHO 1997; Rosen F 1993; Puck JAMA 1997; Curr Opin Immunol 1998; 10:399-406Abbas A et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007Defectos en la Maduración delinfocitos BMéd<strong>ula</strong> óseaGanglio linfáticoActivaciónBtk oµ--, λHipogammaglobulinemiainfantil ligada al X (Bruton)(ausencia de linfocitos B yamígdalas)Algunos pacientes desarrollanenfermedades autoinmunesDefecto en la maduración de Bmas allá del estadio pre-BBtk traduce señales del receptorpre-B
Defectos en la maduración delinfocitos T• Síndrome de DiGeorge: Desarrollodefectuoso del timo, paratiroides yotras estructuras (3ra y 4ta bolsabranquial)– Hipoplasia del timo, maduracióndeficiente de T, alteración enhomeostasis del Ca (tetania), desarrolloanormal de grandes vasos– Deleción en cromosoma 22q11.2
Abbas A et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007Clasificación de lasinmunodeficiencias• Por defectos en la maduración delinfocitos• Por defectos en la activación yfunción de linfocitos• Asociadas con otras enfermedadeshereditarias• Defectos en la inmunidad innata• Defectos en Inmunorreg<strong>ula</strong>ción
Inmunodeficiencias por defectosen activación y función linfocitaria• Deficiencia selectiva de isotipos deIgs: la más común es la de IgA (1:700)• Esporádica, autosómica recesiva odominante• Pacientes pueden ser normales aunquefrecuentemente presentaninfecciones respiratorias y diarreas• Bloqueo en la diferenciación de B encél<strong>ula</strong>s plasmáticas productoras deIgA (probable problema en Th)
Respuesta inmune: CD40/CD40LLinfocito BIgMApoptosisAntígenoLinfocito TCambio de isotipoCD40LCél<strong>ula</strong>s dendríticasRescateAg + CD40Hipermutaciones somáticasMaduración de la afinidadCambio deisotipo ysobrevivienciade altaafinidadIL-<strong>12</strong>
Linfocito BDefecto en Activación de B dependiente de TIgMSíndrome de Hiper-IgMLigado a XMutaciones en CD40LMuestran defectos en inmunidad cel<strong>ula</strong>r (suscep. a Pneumocystis carinni)ApoptosisCambio de isotipoRescateAg + CD40Hipermutaciones somáticasMaduración de la afinidadCambio deisotipo ysobrevivienciade altaafinidadAntígenoCD40LCél<strong>ula</strong>s dendríticasLinfocito TIL-<strong>12</strong>
Alzueta Allergol et Immunopathol 2001: 101; Simonte et al Clin Immunol 2003 (en prensa); Engel et al NatRev Immunol 2003; 3: 813Defecto en diferenciación de B• Inmunodeficiencia comúnvariable:– Producción defectuosa deanticuerpos (por lo menos dosIg: IgG e IgA o IgM)– Disfunción de cél<strong>ula</strong>s B y/o Tde memoria– Se asocia con enfermedadinflamatoria crónica(enfermedad pulmonar crónica,granulomas, enfermedadinflamatoria intestinal)– Esporádica y familiar.Mutaciones en CD40L, ICOS
Defectos en activación y funciónde T: CELULARES Y/O COMBINADAStransmisión de señalesa través del TCRZAP70Ausencia de MHC-II(CIITA, RFX5)Ausencia de MHC-I(defecto de TAP1 yTAP2)Ausencia de CD40LJAK3Defectos en TCR,Mutaciones en ZAP70Defectos en Factores detranscripciónProteína intracel<strong>ula</strong>rinvolucrada en la laseñalización a través delIL2R
Gadola Clin Exp Immunol 2000;<strong>12</strong>1:173; Reith et al Ann Rev Immunol 2001; 19:331; Simonte etal Clin Immunol 2003Defecto en expresión de MHC• Síndrome dellinfocitodesnudo (ClaseII):Mutaciones enRFX5, CIITA• Clase I• Deficiencia deTAP• Deficienciaselectiva deCD8• Ulcerasnecróticas
Abbas A et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007Clasificación de lasinmunodeficiencias• Por defectos en la maduración delinfocitos• Por defectos en la activación yfunción de linfocitos• Asociadas con otras enfermedadeshereditarias• Defectos en la inmunidad innata• Defectos en Inmunorreg<strong>ula</strong>ción
Enfermedades que involucransistemas orgánicos múltiples• Síndrome de Wiskott-Aldrich (Ligada al X)– Eczema, trombocitopenia y susceptibilidad ainfecciones bacterianas– Imposibilidad para producir anticuerpostimodependientes, progresa a disminución delinfocitos e inmunodeficiencia• Ataxia telangiectasia (autosómico recesivo)– Ataxia, malformaciones vasc<strong>ula</strong>res, trastornosneurológicos, tumores, inmunodeficiencia(combinada)
Abbas A et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007Clasificación de lasinmunodeficiencias• Por defectos en la maduración delinfocitos• Por defectos en la activación yfunción de linfocitos• Asociadas con otras enfermedadeshereditarias• Defectos en la inmunidad innata• Defectos en Inmunorreg<strong>ula</strong>ción
Defectos en cél<strong>ula</strong>s fagocíticas y delos componentes del complementoNeutropenia congénita se<strong>ve</strong>ra yneutropenia cíclicaSusceptibilidad a infeccionesbacterianas se<strong>ve</strong>ras y micóticas.Detención en maduración mieloide(promielocito-mielocito)Abbas A et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007
Puck. Puck. JAMA JAMA 1997;278:1835• Enfermedad granulomatosacrónica– Ligada al cromosoma X– Autosómica recesiva o dominante• Adhesión de leucocitos– Defecto de CD18b y/o CD11,CD11c
Puck.Puck.JAMAJAMA1997;278:1835;1997;278:1835;AbbasAbbasA etetalalImmunolImmunolMolMolCelCel200<strong>12</strong>001• Enfermedad granulomatosacrónica– Ligada al cromosoma X– Autosómica recesiva odominante*LPYP ** * ***AP16 años 14 añosYP SP13 añosPaciente<strong>12</strong> años 10 años*(*) Heterocigotopara la mutación (**) ; Homocigoto para la mutación*Niño P **4 años 15 meses,actualmenteasintomático a pesarde producción desuperóxido negativo*Fallecieron entre los 3 y5 años de edad porenfermedadesrespiratoriasGPEVHGRVJV IV82 años 70 añosPV85 añosGG32 añosAVVV EV SV52 años 45 años 43 añosNV42 añosCVCVMV40 años 32 años 28 añosSV35 añosHVMurieron a los 3mesesMurieron a los2 mesesAV14 añosHV<strong>12</strong> añosJV10 añosPV8 años NV19 añosZV2 añosNV5 añosNV13 añosNV11 años
• Defecto de la adhesión deleucocitos (LAD)– Defecto de CD18b y/o CD11,CD11c, compromiso de NK,ausencia del ligando E-selectina
Sindrome de Chediak-Higashi• Autosómico recesivo, muy raro, solo un “cluster” depacientes descritos en los Andes <strong>ve</strong>nezolanos,asociado a tasas elevadas de consanguinidad• Infecciones bacterianas recurrentes, albinismo,trastornos neurológicos• Gránulos gigantes en citoplasma de linfocitosneutrofilos, plaquetas• Fusion incrementada de gránulos citoplasmáticos• Defectos en transporte afectando lisosomas
Inmunodeficiencias primarias:Defectos del complemento• Incremento en la susceptibilidaddesórdenes reumáticos, infección yangioedema.– Componente temprano: (C1, C4, C2) infecciónbacteriana recurrente (gram-positvos)– C3: infección bacteriana recurrente y Neisseriasp– Componente lítico (C5-C9): infección porNeisseria sp
Abbas A et al al Immunol Cell Mol 2005, Marodi L and Notarangelo L. L. Nature Review Immunology, (7), 2007Clasificación de lasinmunodeficiencias• Por defectos en la maduración delinfocitos• Por defectos en la activación yfunción de linfocitos• Asociadas con otras enfermedadeshereditarias• Defectos en la inmunidad innata• Defectos en Inmunorreg<strong>ula</strong>ción
Marodi L and Notarangelo L. Nature Review Immunology, (7), 2007Defectos primarios en inmunorreg<strong>ula</strong>ción:afectan inmunidad innata y adaptativa• Síndromes linfoproliferativos ligados a X– Suscept. a infección por EBV, defectos en citotoxicidaden T y NK, depleción de NKT, alteración en cambio deisotipo de IgG.– Deficiencia de XIAP (incremento en apoptosis)• Síndrome autoinmune linfoproliferativo– Linfadenopatía y hepatoesplenomegalia (no maligno),alteración en apoptosis. Puede cursar con:Trombocitopenia, vasculitis, anemia hemolítica,glomerulonefritis. Hiperganmaglobulinemia. Mutaciones enCD95 (tipo I), CD95L (tipo II), Caspoasa 10 (tipo II)
Marodi L and Notarangelo L. Nature Review Immunology, (7), 2007Defectos primarios en inmunorreg<strong>ula</strong>ción:afectan inmunidad innata y adaptativa• Síndrome parecido a IPEX autosómico recesivo– Deficiencia de CD25: infiltración linfocítica masiva enpuilmones, hígado, bazo, nódulos linfáticos, méd<strong>ula</strong> ósea.Respuesta de T pobre (CD3, PHA, IL-2)– Fenotipicamente similar al IPEX (poliendocrinopatía yenteripatía) que es causado por mutaciones en FOXp3. No hayproducción de IL10 (CD4)• Enfermedad <strong>ve</strong>no-oclusiva hepática con inmunodeficiencia– Causado por oclusión hepática y fibrosis posterior a transplantehematopoyético con daño tóxico al endotelio y a los sinusoideshepáticos. Asociado con linfopenia e hipoganmaglobulinemia yreducido número de T y B
Evaluación sistemática de lasinmunodeficiencias primarias
Evaluación de la historia clínica sugestiva de inmunodeficienciaIdentificación del agente etiológicosi el paciente está infectadoCultivosSerologíaPruebasmolec<strong>ula</strong>resH. influenzaeS. aureusNeumococoAdenovirusEchovirusP. cariniiCandida spM. tuberculosisCitomegalovirusHerpes simplexVaricela-zosterNeisseria spBacterias piógenasS. epidermidisS. marcencensAspergillus spNocardia spCandida spDeficienciahumoralDeficienciacel<strong>ula</strong>rDeficiencias delComplementoDeficiencias de lascél. fagocíticas
Arch Ven Puer Ped 1997; Clin Immunol Immunopathol 1998; 86:237-245Evaluación de un paciente conInmunodeficienciaPruebas de exploración inicialde la función inmuneRecuento diferencial delas cél<strong>ula</strong>s sanguíneasy frotisPrueba DTHCuantificación de lasInmunoglobulinas,electroforesise inmunoelectroforesisFunción fagocítica y SO-CH50 yvia alternaLa asociaciónentre los resultadosobtenidos enambos gruposde pruebas, conduciráa la ejecuciónde otras másespecíficas orientadasal tipo dedefecto encontrado