

Ergebnisse und Diskussion 102 Untersuchung und ... - tuprints
Ergebnisse und Diskussion 102 Untersuchung und ... - tuprints
Ergebnisse und Diskussion 102 Untersuchung und ... - tuprints

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
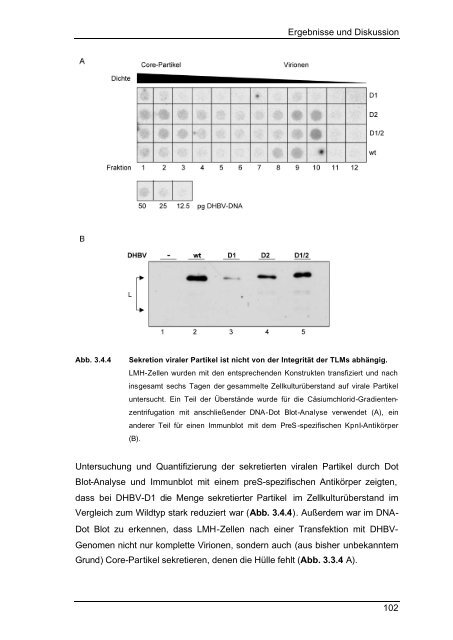
Abb. 3.4.4 Sekretion viraler Partikel ist nicht von der Integrität der TLMs abhängig.<br />
LMH-Zellen wurden mit den entsprechenden Konstrukten transfiziert <strong>und</strong> nach<br />
insgesamt sechs Tagen der gesammelte Zellkulturüberstand auf virale Partikel<br />
untersucht. Ein Teil der Überstände wurde für die Cäsiumchlorid-Gradienten-<br />
zentrifugation mit anschließender DNA-Dot Blot-Analyse verwendet (A), ein<br />
anderer Teil für einen Immunblot mit dem PreS -spezifischen KpnI-Antikörper<br />
(B).<br />
<strong>Untersuchung</strong> <strong>und</strong> Quantifizierung der sekretierten viralen Partikel durch Dot<br />
Blot-Analyse <strong>und</strong> Immunblot mit einem preS-spezifischen Antikörper zeigten,<br />
dass bei DHBV-D1 die Menge sekretierter Partikel im Zellkulturüberstand im<br />
Vergleich zum Wildtyp stark reduziert war (Abb. 3.4.4). Außerdem war im DNA-<br />
Dot Blot zu erkennen, dass LMH-Zellen nach einer Transfektion mit DHBV-<br />
Genomen nicht nur komplette Virionen, sondern auch (aus bisher unbekanntem<br />
Gr<strong>und</strong>) Core-Partikel sekretieren, denen die Hülle fehlt (Abb. 3.3.4 A).<br />
<strong>102</strong>
Tabelle 4.1 Sekretion DNA-haltiger viraler Partikel<br />
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Virus DNA-Menge (in pg) Virale Partikel Virale Partikel per ml<br />
D1 100 3 x 10 7 1,2 x 10 7<br />
D2 425 12,7 x 10 7 5,1 x 10 7<br />
D1/2 462,5 13,9 x 10 7 5,5 x 10 7<br />
wt 300 9 x 10 7 3,6 x 10 7<br />
Im Gegensatz zu DHBV-D1 konnte bei DHBV-D1/2 oder der Mutante DHBV-D2<br />
kein negativer Effekt auf die Sekretion nachgewiesen werden. Dies könnte<br />
bedeuten, dass nicht die TLM-Defizienz per se der Gr<strong>und</strong> für den Sekretions-<br />
defekt von DHBV-D1 war, da diese Mutation auch in der Doppelmutante<br />
vorhanden war. Allerdings könnte es auch sein, dass die TLM-Defizienz von<br />
DHBV-D1 sehr wohl für die Sekretionsdefizienz verantwortlich war, dieser Effekt<br />
jedoch in der Doppelmutante durch eine kompensatorische Mutation im TLM2<br />
aufgehoben wurde. Gr<strong>und</strong>legend könnte auch ein Replikationsdefekt für die<br />
stark verminderte Sekretion verantwortlich sein. Aus diesem Gr<strong>und</strong> wurde die<br />
Mutante DHBV-D1 in folgenden Versuchen nicht eingesetzt. Insgesamt ließ sich<br />
sagen, dass die TLM-Integrität wahrscheinlich für die Sekretion kompletter<br />
viraler Partikel nicht notwendig ist, da DHBV-D2 <strong>und</strong> DHBV-D1/2 eine normale<br />
Sekretion DNA-haltiger Partikel <strong>und</strong> SVPs erlaubten.<br />
Um die Replikationskompetenz der Genome zu überprüfen, wurden die<br />
replikativen Intermediate durch Southern Blot <strong>und</strong> die Core-Expression durch<br />
Immunblot für das wt-Virus <strong>und</strong> die TLM-Mutanten untersucht.<br />
Abb. 3.4.5 Virale Replikation benötigt keine intakten TLMs.<br />
Die DNA der mit den angegebenen Konstrukten transfizierten LMH-Zellen<br />
wurden eine Woche nach Transfektion isoliert <strong>und</strong> im Southern Blot analysiert<br />
103
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
(A) <strong>und</strong> die Proteine durch Immunblot nachgewiesen (B). rc (relaxed circular<br />
DNA), oc (open circular DNA).<br />
In Übereinstimmung mit den obigen <strong>Ergebnisse</strong>n zeigte eine Immunblotanalyse<br />
der zellulären Lysate mit einem Core-spezifischen Antikörper, dass reduzierte<br />
Mengen dieses viralen Proteins in den mit dem D1-Genom transfizierten Zellen<br />
produziert wurden (Abb. 3.4.5 B, Spuren 1 bis 3). Ein Southern-Blot zur<br />
Analyse der viralen replikativen Intermediate zeigte vergleichbare Mengen in<br />
den Fällen von wt-DHBV, DHBV-D2 <strong>und</strong> DHBV-D1/2 <strong>und</strong> im Gegensatz dazu<br />
eine reduzierte DNA-Menge im Falle der DHBV-D1 transfizierten Zellen (Abb.<br />
3.4.5 A, Spuren 1 <strong>und</strong> 4).<br />
Diese Daten sprachen gegen einen Sekretionsblock von DHBV-D1, der<br />
zu einer Retention der viralen Partikel in den Zellen <strong>und</strong> somit erhöhten viralen<br />
Protein- <strong>und</strong> DNA-Mengen führen würde. Es schien wahrscheinlicher, dass die<br />
Mutation im D1-Genom zu einer Störung der Replikation führt. Dies konnte aus<br />
der stark reduzierten Menge replikativer Intermediate in den LMH-Zellen <strong>und</strong><br />
der reduzierten Core-Menge geschlossen werden.<br />
Zusammenfassend zeigen die oben beschriebenen <strong>Ergebnisse</strong>, dass<br />
funktionelle TLMs, möglicherweise mit Ausnahme des TLM1, für die virale<br />
Morphogenese <strong>und</strong> Sekretion entbehrlich sind.<br />
4.1.3 Die Integrität des TLM ist essentiell für die Infektiosität von DHBV<br />
Nachdem sichergestellt war, dass bestimmte TLM-defiziente Viren ohne<br />
sichtbare Defekte sekretiert werden können, wurde die Relevanz der TLMs für<br />
den Infektionsprozess untersucht. Primäre Entenhepatozyten wurden mit wt-<br />
DHBV oder den Mutanten DHBV-D2 <strong>und</strong> DHBV-D1/2 (je 100 GE pro Zelle)<br />
infiziert. Aufgr<strong>und</strong> der in vorangehenden Experimenten beobachteten<br />
reduzierten Replikationsrate von DHBV-D1 wurde die Infektiosität dieses Virus<br />
nicht weiter verfolgt. Die produktive Infektion wurde drei Tage nach der<br />
Inokulation durch indirekte Immunfluoreszenzfärbung <strong>und</strong> Immunblot gegen L<br />
untersucht.<br />
104
Abb. 3.4.6 Inaktivierung der TLMs führt zu einem Infektiositätsverlust.<br />
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
PDHs wurden mit den rekombinanten Viren (100 MGE) infiziert <strong>und</strong> drei Tage<br />
später die Etablierung einer produktiven Infektion mit Hilfe indirekter<br />
Immunfluoreszenzfärbung (A) <strong>und</strong> Immunblot (B) für das virale L-Protein<br />
untersucht.<br />
Die Immunfärbung der Kulturen mit einem L-spezifischen Antikörper zeigte,<br />
dass nur im Falle der mit wt-DHBV infizierten Kulturen de novo-Synthese des<br />
viralen Oberflächenproteins nachgewiesen werden konnte (Abb. 3.4.6 A).<br />
Dieses Ergebnis wurde durch die Immunblotanalyse der zellulären Lysate mit<br />
einem L-spezifischen Antikörper bestätigt (Abb. 3.4.6 B). Im Falle der wt-<br />
infizierten Zellen waren signifikante Signale nachweisbar, während in den<br />
Lysaten der mit DHBV-D2 oder DHBV-D1/2-infizierten Zellen keine L-<br />
spezifischen Signale sichtbar waren, die auf eine produktive Infektion<br />
hingedeutet hätten. Dies könnte entweder dadurch bedingt sein, dass der<br />
verwendete Antikörper die mutierten L-Proteine schlecht erkannte oder dass<br />
tatsächlich sehr wenig L-Protein vorhanden war. Um daher die Etablierung<br />
einer Infektion direkt zu analysieren, wurde die cccDNA-Etablierung <strong>und</strong><br />
Amplifikation in den infizierten Zellen untersucht.<br />
105
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Die Etablierung einer produktiven Infektion wird durch die Bildung von<br />
cccDNA während der Infektion charakterisiert. Die Analyse der cccDNA-Menge<br />
fünf Tage nach der Infektion durch eine PCR, die präferentiell cccDNA<br />
amplifiziert, zeigte, dass nur im Falle der mit wt-DHBV-infizierten Kulturen<br />
cccDNA nachweisbar war (Abb. 3.4.7). Nach einer Infektion mit TLM-<br />
defizienten Viren wurde die eventuell aufgenommene rcDNA nicht in cccDNA<br />
konvertiert <strong>und</strong> so keine Infektion etabliert. Dies bestätigte die oben gezeigten<br />
Daten des Immunblots <strong>und</strong> der Fluoreszenzfärbung (Abb. 3.4.6).<br />
Abb. 3.4.7 Keine cccDNA-Bildung nach Infektion mit TLM-defizienten Viren.<br />
PDHs wurden mit den rekombinanten Viren infiziert <strong>und</strong> nach fünf Tagen<br />
wurden die Kulturen für die cccDNA-Analyse geerntet. cccDNA wurde isoliert,<br />
ein Verdau mit Plasmid Safe-DNase durchgeführt <strong>und</strong> eine PCR durchgeführt,<br />
die präferentiell diese virale DNA -Spezies amplifiziert.<br />
Zusammengenommen zeigten diese Daten, dass die Zerstörung von TLM2<br />
oder beider TLMs die Infektiosität von DHBV stark reduziert. Die Bildung der<br />
cccDNA nach Inokulation der Zellen mit TLM-defizienten Viren ist gestört.<br />
4.1.4 Zerstörung der TLMs beeinflusst weder Bindung noch Internalisierung der<br />
viralen Partikel<br />
Um zu untersuchen, ob der Infektiositätsverlust der TLM-Mutanten auf eine<br />
reduzierte Bindung an oder gestörte Internalisierung der Viren in die Wirtszellen<br />
zurückzuführen ist, wurde ein Bindungs- <strong>und</strong> Internalisierungsassay<br />
durchgeführt. Die primären Leberzellkulturen wurden für 2 h bei 4°C mit 100<br />
MGE inkubiert, dann wurden die Kulturen entweder nach Waschen direkt<br />
geerntet oder auf 37°C erwärmt, um die Aufnahme der Viren zu erlauben. Diese<br />
Proben wurden mit Trypsin geerntet, um nur intrazelluläres Virus<br />
106
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
nachzuweisen. Alle Ansätze wurden erst mit Proteinase K verdaut <strong>und</strong> dann<br />
eine DHBV-DNA-spezifische PCR durchgeführt.<br />
Abb. 3.4.8 Zerstörung der TLMs beeinflusst weder Bindung noch Internalisierung der<br />
viralen Partikel.<br />
Mit den rekombinanten Viren wurde ein Bindungs- <strong>und</strong> Internalisierungsassay<br />
durchgeführt. Anschließend wurden die Zellen geerntet, mit Proteinase K<br />
verdaut <strong>und</strong> eine PCR durchgeführt.<br />
Dieser Assay zeigte, dass ähnliche Mengen Virus-DNA-haltiger Partikel nach<br />
Inokulation mit wt-DHBV oder den Mutanten an die Zellmembran binden (Abb.<br />
3.4.8, Spuren 1 bis 4, oben). Darüber hinaus zeigte eine Analyse der<br />
intrazellulären viralen DNA 3 h nach Beginn des Viruseintritts in die Zellen nur<br />
leichte Unterschiede zwischen wt-Virus <strong>und</strong> den TLM-Mutanten (Abb. 3.4.8,<br />
Spuren 1 bis 4, unten). DHBV-D1/2 schien etwas weniger aufgenommen zu<br />
werden als das wt-Virus <strong>und</strong> DHBV-D2; dieser leichte Unterschied würde<br />
jedoch nicht den drastischen Infektiositätsverlust dieser Mutante erklären.<br />
Die Daten weisen darauf hin, dass der Verlust der Infektiosität nach<br />
Inaktivierung der TLMs weder auf eine verminderte Bindung noch auf eine<br />
Beeinträchtigung der sehr frühen Infektionsschritte zurückzuführen ist. Der<br />
Infektiositätsverlust der TLM-defizienten Viren muss damit auf die Inaktivierung<br />
eines Schrittes nach Bindung <strong>und</strong> Internalisierung zurückzuführen sein.<br />
107
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
4.1.5 Die Integrität des TLM ist für den Austritt von DHBV aus Endosomen<br />
essentiell<br />
Die oben beschriebenen <strong>Ergebnisse</strong> zeigten, dass die Zerstörung der TLMs die<br />
Infektiosität von DHBV stark herabsetzt, ohne die virale Bindung oder<br />
Aufnahme zu beeinflussen. Unter der Annahme, dass DHBV nach Bindung an<br />
einen Rezeptor oder Rezeptorkomplex durch Rezeptor-vermittelte Endozytose<br />
aufgenommen wird [60, 81], wurde die Hypothese aufgestellt, dass die<br />
Zerstörung der TLMs in der Akkumulation des Virus in endosomalen<br />
Kompartimenten resultieren könnte. In diesem Modell wären beide TLMs,<br />
ähnlich zu den bei anderen Viren beschriebenen Fusionspeptiden, daran<br />
beteiligt, das Virus aus dem endosomalen Kompartiment freizusetzen. Um dies<br />
zu untersuchen, wurden subzelluläre Kompartimente 9 h nach Inokulation der<br />
Zellen mit den rekombinanten Viren durch differentielle Zentrifugation aus PDHs<br />
isoliert <strong>und</strong> in der endosomalen sowie zytosolischen Fraktion in Berlin durch<br />
Taqman-PCR die Menge der DHBV-DNA analysiert.<br />
Abb. 3.4.9 TLM-Defizienz führt zur verminderten Freisetzung viraler DNA in das<br />
Zytosol.<br />
PDHs wurden mit 400 MGE der rekombinanten Viren inokuliert <strong>und</strong> 9 h bei<br />
37°C inkubiert. Anschließend wurden die Zellen mit Trypsin geerntet <strong>und</strong> eine<br />
108
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
subzelluläre Fraktionierung durchgeführt. Danach wurde die virale DNA aus den<br />
Proben isoliert <strong>und</strong> durch Taqman-PCR am Robert-Koch-Institut in Berlin<br />
bestimmt. SA (Standardabweichung), e (endosomale Fraktion), c (zytosolische<br />
Fraktion). Abb. zur Verfügung gestellt von E. Hildt (RKI, Berlin).<br />
9 h nach der Inokulation zeigte die PCR ähnliche Mengen DHBV-DNA in der<br />
endosomalen Fraktion von Zellen, die mit DHBV-D1, DHBV-D2 <strong>und</strong> DHBV-D1/2<br />
infiziert wurden. Eine kleinere Menge wurde in Zellen gef<strong>und</strong>en, die mit dem wt-<br />
Virus infiziert wurden (Abb. 3.4.9). Im Gegensatz dazu zeigte sich nur im Falle<br />
der mit wt-Virus infizierten Zellen ein signifikantes Signal im Zytosol. Es wurden<br />
keine signifikanten Mengen DNA in der zytosolischen Fraktion der mit den<br />
Mutanten infizierten Zellen nachgewiesen (Abb. 3.4.9). Dies deutete darauf hin,<br />
dass die TLM-defizienten Viren zwar in endosomale Kompartimente<br />
aufgenommen werden, jedoch im Gegensatz zum wt-Virus daraus nicht<br />
freigesetzt werden können.<br />
Die <strong>Ergebnisse</strong> weisen darauf hin, dass die Funktionalität der TLM<br />
Voraussetzung für die Freisetzung internalisierter viraler Partikel aus dem<br />
Endosom in das Zytosol ist.<br />
4.2 <strong>Diskussion</strong><br />
Die Entdeckung des TLMs in der preS-Domäne des L-Proteins von HBV, das<br />
Energie- <strong>und</strong> Rezeptor-unabhängig in Form eines Peptids durch Membranen<br />
translozieren kann [181], führte zur <strong>Untersuchung</strong> der Rolle dieses Motivs im<br />
Lebenszyklus von Hepadnaviren.<br />
Eine Datenbankanalyse, die in Berlin am RKI von E. Hildt durchgeführt<br />
wurde <strong>und</strong> auf den Parametern der amphipatischen Helix basierte, ergab, dass<br />
die Oberflächenproteine aller bekannten Hepadnaviren putative TLMs in der<br />
preS-Domäne enthalten. Auffallend war dabei, dass DHBV im Gegensatz zu<br />
HBV <strong>und</strong> den anderen Hepadnaviren anscheinend zwei TLMs besitzt. Dies<br />
könnte durch die unterschiedliche Organisation der Gene für die<br />
Oberflächenproteine im Vergleich zu HBV bedingt sein. HBV besitzt die drei<br />
Oberflächenproteine SHBs, MHBs <strong>und</strong> LHBs, wobei das TLM in MHBs <strong>und</strong><br />
LHBs vorkommt. DHBV besitzt nur die beiden Hüllproteine L <strong>und</strong> S, wobei die<br />
109
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
TLMs nur im L vorkommen. Der Vollständigkeit halber soll jedoch darauf<br />
hingewiesen werden, dass aminoterminal verkürzte L-Proteine in geringer<br />
Konzentration auch von DHBV exprimiert werden [184]. Keines dieser<br />
verkürzten L-Proteine ist für die DHBV-Infektion essentiell [185].<br />
Die Funktionalität der vorhergesagten TLMs wurde durch einen Assay in<br />
Berlin bestätigt (Abb. 3.4.2). Dieser zeigte, dass die vorhergesagten TLMs eine<br />
vergleichbare Zellpermeabilität zu dem zuerst identifizierten TLM aus dem<br />
humanen Prototyp-Virus HBV aufweisen.<br />
Die beobachtete Konservierung der TLMs in den Oberflächenproteinen<br />
der verschiedenen Hepadnaviren deutet auf eine wichtige Rolle im viralen<br />
Lebenszyklus hin. Es wurde jedoch keine Beeinträchtigung der Morphogenese<br />
oder Sekretion viraler Partikel nach einer funktionellen Inaktivierung der TLMs<br />
beobachtet. Dies stimmt mit dem aktuellen Modell der HBV-Morphogenese<br />
überein, das keine Membrantranslokation beschreibt, sondern Budding-<br />
Prozesse, die keine Zellpermeabilität benötigen [186]. Die reduzierte<br />
Replikation im Falle von DHBV-D1 scheint auf einen sek<strong>und</strong>ären Effekt<br />
zurückzuführen sein, da die Doppelmutante, die dieselbe Mutation enthält,<br />
keinen Replikationsdefekt zeigte. Allerdings könnte die zweite Mutation in<br />
DHBV-D1/2 auch einen kompensatorischen Effekt auf die erste haben.<br />
Im Gegensatz zur Morphogenese scheint die Infektiosität durch die<br />
Inaktivierung der TLMs stark beeinflusst zu werden, die eingeführten<br />
Mutationen führten zu einem nahezu kompletten Verlust der Infektiosität. Dieser<br />
wurde nicht durch eine reduzierte Bindung oder Aufnahme der DHBV-Mutanten<br />
in die Wirtszellen verursacht. Das bedeutet, dass die veränderte Struktur des<br />
Oberflächenproteins der rekombinanten Viren keinen Einfluss auf die<br />
Rezeptorbindung hat. Es kann jedoch nicht ausgeschlossen werden, dass die<br />
Bindung <strong>und</strong> die darauf folgende Internalisierung bei diesen Mutanten sich vom<br />
normalen Aufnahmeweg des DHBV unterscheidet. Dies scheint jedoch<br />
unwahrscheinlich, da die subzelluläre Fraktionierung sowohl das wt-Virus als<br />
auch die Mutanten in denselben Zellkompartimenten offenbarte. Insgesamt<br />
zeigt dies, dass die TLM-Strukturen sowohl für die virale Bindung als auch<br />
Internalisierung entbehrlich sind. Das deutet auf einen Infektionsblock nach der<br />
viralen Aufnahme hin.<br />
110
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Da die TLM-defektiven Viren in die Zellen aufgenommen wurden, jedoch<br />
keine cccDNA gebildet wurde, kann gefolgert werden, dass der Infektionsblock<br />
der Viren auf dem Weg von der Zelloberfläche zum Nukleus liegt, denn nur die<br />
rcDNA eintretender Viren, die zum Nukleus gelangt, kann in cccDNA<br />
umgewandelt werden.<br />
Werden die viralen Partikel durch einen endozytischen Weg in die<br />
Wirtszellen aufgenommen, liegen sie nach der Aufnahme in Endosomen vor.<br />
Da der endosomale Aufnahmeweg oft zum Abbau der aufgenommenen<br />
Substanzen führt, muss das aufgenommene Virus aus diesem Weg austreten.<br />
In Endosomen könnten die TLMs nach einem bestimmten Signal exponiert<br />
werden. Dies könnte zur Freisetzung der Viren aus dem endosomalen<br />
Kompartiment führen. Die Exposition eines Fusionssignals während der<br />
Infektion von Wirtszellen wurde für andere Viren beschrieben. So haben z.B.<br />
Homotrimere des Hämagglutinins von Influenza-Viren fusogene Aktivität (als<br />
HA0-Vorläuferproteine). Nach einer endolytischen Spaltung durch eine Furin-<br />
ähnliche Protease entstehen HA1 <strong>und</strong> HA2. Dieser Spaltungsschritt ermöglicht<br />
die HA-vermittelte Fusion durch Freisetzung bestimmter Sequenzen, die durch<br />
Konformationsveränderungen Membranfusionen induzieren können [133]. Im<br />
Falle des Poliovirus führt die Interaktion des Virus mit seinem Rezeptor zur<br />
Freisetzung des N-myristylierten VP4-Peptids <strong>und</strong> der Exposition einer<br />
amphipatischen Sequenz von VP1, die in die Wirtsmembran inseriert <strong>und</strong> so<br />
den Eintritt des Virus in das Zytoplasma ermöglicht [187].<br />
Interessanterweise benötigt die Infektiosität von DHBV die Integrität<br />
beider TLMs. Ein Defekt in TLM2 kann nicht durch ein intaktes TLM1<br />
kompensiert werden. Experimente mit zellpermeablen Kapsiden mit TLM-<br />
HBcAg-Fusionsproteinen zeigten, dass die Zellpermeabilität dieser Partikel<br />
durch die Verwendung zweier TLMs, die in einem ähnlichen Abstand liegen wie<br />
im preS-Teil des DHBV-L, im Vergleich zu nur einem TLM stark verbessert wird<br />
(B. Brandenburg, persönliche Mitteilung). Dies deutet darauf hin, dass beide<br />
TLMs für eine effiziente Permeation benötigt werden <strong>und</strong> einen synergistischen<br />
Effekt ausüben.<br />
Die oben beschriebenen <strong>Ergebnisse</strong> könnten durch einen ähnlichen<br />
Mechanismus bei Hepadnaviren erklärt werden. Nach einer Rezeptor-<br />
vermittelten Endozytose des viralen Partikels <strong>und</strong> dem Eintritt in den<br />
111
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
endosomalen Transportweg findet eine proteolytische oder pH-induzierte<br />
Prozessierung des viralen Oberflächenproteins statt. Diese Prozessierung<br />
könnte zu veränderten viralen Partikeln führen, die an der Oberfläche<br />
exponierte TLMs besitzen. Diese TLMs könnten dann den Austritt des Virus aus<br />
dem Endosom ermöglichen, bevor es zu einer Inaktivierung des Virus in einem<br />
lysosomalen Kompartiment kommt.<br />
In zukünftigen Studien sollte der genaue Mechanismus der<br />
Virusfreisetzung aus dem endosomalen Kompartiment <strong>und</strong> der Beitrag der<br />
TLMs dabei untersucht werden. Sollten die TLMs wie oben beschrieben nach<br />
einer Proteolyse exponiert werden, könnte man nach der Exposition der Viren<br />
mit endosomalen Kompartimenten eine Immunpräzipitation mit einem<br />
Antikörper durchführen, der die TLMs spezifisch bindet. Außerdem sollte<br />
untersucht werden, ob eine Translokation der Viren aus den Endosomen in das<br />
Zytosol stattfindet, bei der die virale Membran erhalten bleibt oder ein normaler<br />
Fusionsvorgang vorliegt, bei dem die virale mit der Wirtsmembran fusioniert.<br />
112
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
5. Die Ausbreitung von Hepadnaviren erfolgt in vitro durch<br />
extrazelluläre Nachkommensviren<br />
Wenn Entenküken mit einer niedrigen MOI mit DHBV infiziert werden, sind<br />
innerhalb kurzer Zeit (etwa eine Woche) alle Hepatozyten in der Leber infiziert<br />
[188]. Die Art der viralen Ausbreitung muss sehr effizient sein, besonders wenn<br />
man die Größe der Leber mit etwa 10 11 Hepatozyten berücksichtigt. In vivo<br />
werden oft Gruppen Virus-replizierender Zellen in der frühen Phase einer<br />
Infektion beobachtet [189]. Zusätzlich können ähnliche Cluster auch in<br />
Primärkulturen von Hepatozyten beobachtet werden (eigene Beobachtung <strong>und</strong><br />
die anderer Arbeitsgruppen). Zusammengenommen legen diese<br />
Beobachtungen indirekt nahe, dass Hepatitis B-Viren direkt von einer infizierten<br />
Zelle auf eine nicht-infizierte Zelle übertragen werden <strong>und</strong> deuten auf eine<br />
Bewegung der Nachkommensviren außerhalb der infizierten Zelle hin, die nicht<br />
nur durch Diffusion kontrolliert sein kann. Auf der anderen Seite ist bekannt,<br />
dass eine große Menge von Virionen <strong>und</strong> subviralen Partikeln von infizierten<br />
Zellen in den extrazellulären Raum sekretiert werden. Dort sollte freie Diffusion<br />
die Infektion weit entfernt gelegener Zellen ermöglichen. Um solide<br />
Informationen über die Art der viralen Ausbreitung innerhalb einer primären<br />
Leberzellkultur zu erhalten, wurden die folgenden <strong>Untersuchung</strong>en<br />
durchgeführt.<br />
Es wurde analysiert, ob bei Hepatitis B-Viren für die Übertragung ihrer<br />
Nachkommensviren von einer infizierten auf eine nicht-infizierte Zelle nur<br />
intrazelluläres Virus verantwortlich ist oder ob die Sekretion von<br />
Nachkommensviren in den extrazellulären Raum zur Verbreitung der Infektion<br />
beiträgt. Im ersten Fall sollte die Ausbreitung der Infektion in einer Zellkultur<br />
nicht durch den Einsatz neutralisierender Antikörper oder Substanzen, die die<br />
Bindung der Viren an ihre Zielzellen hemmen, beeinträchtigt werden. Die<br />
intrazellulären Viren wären vor diesen Substanzen geschützt <strong>und</strong> bräuchten<br />
außerdem nicht an die Oberfläche einer Zielzelle zu binden, um diese zu<br />
infizieren. Für die <strong>Untersuchung</strong>en wurden verschiedene neutralisierende<br />
Antiseren [190, 191] sowie die synthetische Substanz Suramin eingesetzt. Wie<br />
in Kapitel III.3.1 gezeigt, vermindern diese Substanzen die Bindung viraler<br />
113
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Partikel <strong>und</strong> die anschließende Infektion stark <strong>und</strong> waren damit geeignet, die<br />
Ausbreitung von Hepadnaviren in Zellkultur zu untersuchen.<br />
5.1 <strong>Ergebnisse</strong><br />
5.1.1 Neutralisierende Antikörper <strong>und</strong> Suramin verhindern die virale<br />
Ausbreitung in vitro<br />
PDHs wurden mit DHBV infiziert, <strong>und</strong> drei Tage später wurde dem<br />
Zellkulturüberstand Suramin oder preS-Antiserum zugegeben. Am Tag 3 war<br />
die produktive DHBV-Infektion in den Hepatozyten etabliert <strong>und</strong> die ersten<br />
Nachkommensviren sollten freigesetzt werden. Drei Tage nach dem Beginn der<br />
Behandlung wurden die Zellen dann fixiert <strong>und</strong> das L-Protein als Indikator für<br />
die produktive Infektion durch indirekte Immunfluoreszenzfärbung<br />
mikroskopisch analysiert.<br />
114
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Abb. 3.5.1 Neutralisierende Antikörper <strong>und</strong> Suramin verhindern die virale Aus-<br />
breitung in vitro.<br />
PDHs wurden mit DHBV infiziert <strong>und</strong> drei Tage später der neutralisierende<br />
Antikörper DPSI oder Suramin zugegeben, um eine Ausbreitung durch<br />
freigesetzte Viren von den primär infizierten Zellen auf nicht-infizierte Zellen zu<br />
verhindern. Das Medium wurde täglich gewechselt <strong>und</strong> neue Substanzen<br />
zugegeben. Nach drei Tagen wurden die Zellen fixiert <strong>und</strong> die infizierten Zellen<br />
durch Immunfärbung des L-Proteins analysiert. A zeigt eine 10-fache Ver-<br />
größerung der Kulturen, B eine 40-fache.<br />
Die L-Färbung zeigte, dass in infizierten, unbehandelten PDH-Kulturen die<br />
infizierten Zellen nicht zufällig in der Kultur verteilt waren, sondern in Gruppen<br />
angeordnet waren (Abb. 3.5.1 A). In jeder dieser Gruppen waren eine oder<br />
mehrere Hepatozyten am stärksten gefärbt, diese waren von weniger stark<br />
gefärbten Zellen umgeben. Die Behandlung der Kulturen mit neutralisierenden<br />
Antiseren verhinderte diese Gruppenbildung, es konnten hauptsächlich stark<br />
gefärbte Zellen beobachtet werden (Abb. 3.5.1). Suramin hatte denselben<br />
Effekt in den behandelten Kulturen.<br />
Um die Möglichkeit auszuschließen, dass die verwendeten Reagenzien<br />
die virale Ausbreitung verhinderten, indem sie negativ auf die intrazelluläre<br />
virale Replikation oder die Sekretion der Nachkommensviren wirkten, wurde die<br />
persistent DHBV-produzierende LMH-Zelllinie D2 für drei Tage kontinuierlich<br />
mit Suramin oder neutralisierendem preS-Antikörper behandelt. Anschließend<br />
wurden die Kulturen sowie die Zellkulturüberstände geerntet <strong>und</strong> die virale DNA<br />
<strong>und</strong> L-Protein analysiert.<br />
115
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Abb. 3.5.2 preS-Antikörper <strong>und</strong> Suramin beeinträchtigen weder virale Sekretion noch<br />
Replikation.<br />
Die DHBV-replizierende Zelllinie D2 wurde mit dem preS -spezifischen<br />
Antiserum KpnI oder Suramin für drei Tage kontinuierlich behandelt. Danach<br />
wurden die Zellkulturüberstände <strong>und</strong> die Zellen geerntet. (A) PCR-Analyse der<br />
viralen DNA in den Überständen. 5 µl der Überstände wurden in PCR-<br />
Lysepuffer aufgenommen <strong>und</strong> für eine DHBV-DNA-spezifische PCR eingesetzt.<br />
Amplifizierte Produkte wurden auf einem mit Ethidiumbromid gefärbten<br />
Agarosegel nach der Elektrophorese visualisiert. (B) Immunblot-Analyse des<br />
intrazellulären L-Gehalts. Behandelte Zellen wurden in Lämmli-Lysepuffer lysiert<br />
<strong>und</strong> für einen L-Immunblot eingesetzt.<br />
Es konnte weder ein Effekt auf die virale Sekretion (Abb. 3.5.2 A) noch auf die<br />
L-Expression (Abb. 3.5.2 B) beobachtet werden. Dies spricht gegen einen<br />
Effekt der Substanzen auf virale Replikation oder Sekretion.<br />
Die oben beschriebenen Daten zeigen, dass Hepadnaviren von einer<br />
primär infizierten Hepatozyte auf eine nicht-infizierte Zelle übertragen werden,<br />
indem sie von der ersten Zelle in den extrazellulären Raum freigesetzt werden<br />
<strong>und</strong> dann eine andere Zelle infizieren.<br />
Es blieb jedoch offen, warum Gruppen infizierter Zellen beobachtet<br />
wurden. Dies sollte nicht der Fall sein, wenn die Ausbreitung nicht über<br />
Zellverbindungen erfolgt, sondern nur durch sekretierte Nachkommensviren.<br />
Diese könnten an zufälligen Stellen der Plasmamembran einer infizierten Zelle<br />
freigesetzt werden, von wo aus sie ungehindert im extrazellulären Raum<br />
diff<strong>und</strong>ieren oder weggeschwemmt werden können. Eine Erklärung für die<br />
116
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Gruppenbildung könnte eine polarisierte Ausschleusung der Nachkommens-<br />
viren in interzelluläre Kompartimente sein. Diese Kompartimente würden eine<br />
freie Diffusion der Viren verhindern, jedoch zugänglich für neutralisierende<br />
Antikörper <strong>und</strong> Suramin sein. Wenn diese Annahme korrekt ist, so müsste man<br />
annehmen, dass Nachkommensviren nicht an zufälligen Stellen der<br />
Plasmamembran sekretiert werden, sondern präferentiell an spezifischen<br />
Regionen. Dies ist gut vorstellbar, da polarisierter Egress für andere Viren wie<br />
HIV schon gezeigt wurde [192] <strong>und</strong> insbesondere Hepatozyten für ihre stark<br />
polarisierte Organisation bekannt sind [193, 194].<br />
Um einen ersten Hinweis auf den oben genannten Egress <strong>und</strong> den<br />
Modus der sek<strong>und</strong>ären Infektion zu erhalten, wurden elektronenmikroskopische<br />
<strong>und</strong> Immunfluoreszenz-Analysen durchgeführt.<br />
Abb. 3.5.3 Elektronenmikroskopische Aufnahme der intrazellulären Verteilung von<br />
viralen Partikeln in PDHs.<br />
Chronisch infizierte PDHs wurden sieben Tage nach Ausplattierung fixiert <strong>und</strong><br />
für die Elektronenmikroskopie präpariert. Eine Detailansicht des Zytoplasmas<br />
der infizierten Zelle ist gezeigt mit zwei einzelnen Vesikeln (Pfeile), Virionen<br />
(weiße Pfeilspitzen) <strong>und</strong> einer Anzahl von SVPs (offene Pfeilspitzen). Die<br />
Abbildung wurde von H. Hohenberg (Heinrich-Pette-Institut, Hamburg) zur<br />
Verfügung gestellt.<br />
117
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Die ultrastrukturelle Analyse offenbarte, dass Virionen (Abb. 3.5.3, weiße<br />
Pfeilspitzen) <strong>und</strong> subvirale Partikel (Abb. 3.5.3, offene Pfeilspitzen) in<br />
Membran-umgebenen, zytoplasmatischen Vesikeln <strong>und</strong> dort in einigen auch<br />
gemeinsam zu finden sind (Abb. 3.5.3, Pfeile). Wie erwartet sind Virionen in<br />
den Vesikeln zahlenmäßig weit unterrepräsentiert.<br />
Abb. 3.5.4 Konfokale, mikroskopische Analyse der zellulären L-Protein-Verteilung in<br />
PDHs.<br />
Kulturen auf Glasplättchen wurden infiziert <strong>und</strong> sieben Tage später fixiert. Eine<br />
indirekte Immunfluoreszenzfärbung für das virale L-Protein wurde durchgeführt<br />
<strong>und</strong> mit einem konfokalen Laserscanningmikroskop analysiert. Ausgesuchte<br />
Schnitte derselben Gruppe von Zellen (1 bis 6, von apikal nach lateral) sind<br />
gezeigt. Die Pfeilspitzen zeigen die Färbung der Plasmamembran, die Pfeile<br />
zeigen partikelgefüllte Vesikel in den Hepatozyten.<br />
Die L-Immunfärbung infizierter Hepatozyten zeigte ein punktförmiges Muster,<br />
das konsistent mit den elektronenmikroskopischen Aufnahmen war. Es wies<br />
zusätzlich darauf hin, dass präformierte, virale Partikel sub-kompartimentalisiert<br />
in vesikulären Strukturen im Zytoplasma vorkommen (Abb. 3.5.4, Pfeilspitzen).<br />
Die <strong>Untersuchung</strong> einer kleinen Kolonie von infizierten Hepatozyten durch<br />
konfokale Laserscanning-Mikroskopie mit Schnitten von etwa 0,3 µm Dicke<br />
118
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
ergab, dass die L-haltigen Vesikel nicht zufällig im Zytoplasma verteilt sind. Sie<br />
akkumulieren präferentiell an der basolateralen Seite der Zytoplasmamembran<br />
(Abb. 3.5.4, Pfeile). Darüber hinaus war auch eine starke Färbung der<br />
Plasmamembran sichtbar (Abb. 3.5.4, Pfeilspitzen).<br />
5.2 <strong>Diskussion</strong><br />
Wie im vorangehenden Ergebnis-Teil gezeigt, verhinderten sowohl<br />
neutralisierende Antikörper als auch Suramin die Ausbreitung von<br />
Hepadnaviren in Zellkultur. Voraussetzung dafür war, dass diese Substanzen<br />
die Bindung der Viren an ihre Wirtszellen blockierten, jedoch nicht mit späteren<br />
Schritten der Infektion interferierten. Unspezifische Effekte der Substanzen auf<br />
die Infektion konnten ausgeschlossen werden, da eine Behandlung, nachdem<br />
Inokulum zur Zellkultur gegeben wurde, keinen Effekt auf die Infektion hatte. Es<br />
scheint ebenso unwahrscheinlich, dass die Substanzen intrazellulär wirkten, da<br />
in D2-Zellen kein Effekt auf eine bestehende Infektion beobachtet wurde.<br />
Wurden infizierte Zellkulturen nach 6 Tagen fixiert <strong>und</strong> für L gefärbt, so<br />
waren in der Immunfluoreszenzfärbung unterschiedlich stark leuchtende<br />
Hepatozyten erkennbar. Besonders stark leuchtende Zellen waren oft von<br />
weniger stark leuchtenden Zellen umgeben. Erstere exprimierten sehr<br />
wahrscheinlich größere Mengen L als letztere. Sie stellten daher vermutlich die<br />
primär infizierten Zellen dar, von denen die Nachkommensviren produziert <strong>und</strong><br />
benachbarte Zellen infiziert wurden. In enger Nachbarschaft zu diesen Zellen<br />
waren weniger stark gefärbte Hepatozyten sichtbar, die in größerer Zahl als die<br />
stark gefärbten Zellen vorhanden waren. Dies ließ vermuten, dass sie von den<br />
Nachkommensviren der primär infizierten Zellen infiziert wurden. In den mit<br />
Suramin oder neutralisierenden Antikörpern behandelten Zellkulturen fehlten<br />
diese sek<strong>und</strong>är infizierten Hepatozyten weitgehend. Dies deutete darauf hin,<br />
dass sekretierte Nachkommensviren von den neutralisierenden Antikörpern<br />
inaktiviert werden. Sie waren offensichtlich für die Antikörper zugänglich. Das<br />
erhaltene Ergebnis lässt es sehr unwahrscheinlich erscheinen, dass<br />
Hepadnaviren ihre Nachkommensviren durch direkten Transport von infizierter<br />
Zelle zu nicht-infizierter Zelle verbreiten. Die Wirksamkeit von Suramin in diesen<br />
119
<strong>Ergebnisse</strong> <strong>und</strong> <strong>Diskussion</strong><br />
Versuchen zeigt, dass Nachkommensviren sezerniert werden, die anschließend<br />
an die Zelloberfläche nicht-infizierter Zellen binden <strong>und</strong> diese dann infizieren.<br />
Zusammen weisen die Daten darauf hin, dass Hepadnaviren zuerst von<br />
der primär infizierten Hepatozyte in den extrazellulären Raum freigesetzt<br />
werden <strong>und</strong> dann an die Oberfläche einer nicht-infizierten, benachbarten Zelle<br />
binden müssen, um eine erfolgreiche Verbreitung ihrer Nachkommen zu<br />
garantieren. Die elektronenmikroskopischen <strong>und</strong> Fluoreszenzfärbungsanalysen<br />
unterstützen diese Annahmen. Virale Partikel wurden nur in zytoplasmatischen<br />
Vesikeln gef<strong>und</strong>en. Keine einzelnen freien Partikel konnten beobachtet werden.<br />
Diese Partikel könnten leicht durch direkte Zell-Zell-Verbindungen übertragen<br />
werden. Die virushaltigen Vesikel werden wahrscheinlich an die Zellmembran<br />
transportiert, um dort ihren Inhalt freizusetzen. Folglich wurden auch die<br />
meisten Vesikel in Nähe der Zytoplasmamembran infizierter Hepatozyten<br />
gef<strong>und</strong>en. Obwohl einige wenige Vesikel nahe der apikalen Membran<br />
vorhanden waren, waren die meisten in Nähe der basolateralen lokalisiert.<br />
Wenn diese Viruspartikel-gefüllten Vesikel mit der Plasmamembran fusionieren,<br />
würden Virionen <strong>und</strong> subvirale Partikel zusammen <strong>und</strong> gerichtet in den<br />
basolateral lokalisierten, extrazellulären Raum freigesetzt. Dies könnte die<br />
Erklärung dafür sein, dass freie Diffusion der viralen Partikel nicht stattfindet<br />
<strong>und</strong> bevorzugt benachbarte Zellen infiziert werden.<br />
In zukünftigen <strong>Untersuchung</strong>en könnte die genaue Stelle der viralen<br />
Sekretion bestimmt werden. Dazu könnten z.B. Immunfluoreszenzstudien mit<br />
Antikörpern gegen Marker der unterschiedlichen Membranen (z.B. apikal vs.<br />
lateral) durchgeführt werden, die eine präferentielle Lokalisierung der<br />
virushaltigen Vesikel in der Zelle aufzeigen könnten.<br />
120
IV. Material <strong>und</strong> Methoden<br />
1. Material<br />
1.1 Chemikalien<br />
Material <strong>und</strong> Methoden<br />
Sofern nicht anders angegeben, wurden die Chemikalien der Firmen Sigma (Deisenhofen),<br />
Roche Diagnostics (Mannheim), Roth (Karlsruhe), Biozol (München), Serva (Heidelberg) <strong>und</strong><br />
Merck (Darmstadt) verwendet. Radioaktive Reagenzien wurden von der Firma Hartmann<br />
Analytik (Göttingen) bezogen.<br />
Die verwendeten Chemikalien wiesen den Reinheitsgrad ‚reinst’ oder ‚zur Analyse’ auf.<br />
Alle Medien, Puffer <strong>und</strong> Lösungen wurden mit deionisiertem Wasser aus einer Wasser-<br />
aufbereitungsanlage RO 35 TS-System von Millipore angesetzt.<br />
1.2 Häufig verwendete Puffer <strong>und</strong> Lösungen<br />
Blocking-Reagenz 3 % Trockenmilch<br />
500 ml 1 x TBS<br />
0,1 % Tween ® 20<br />
DNA-Probenpuffer 30 % Glyzerin<br />
6 x 60 mM EDTA<br />
6 mM Tris, pH 7<br />
Lämmli-Lysepuffer 100 mM Tris, pH 6,8<br />
[195] 20 % Glyzerin<br />
2 % SDS<br />
0,1 % Bromphenolblau<br />
0,2 % ß-ME<br />
PBS 140 mM NaCl<br />
10 x; pH 7,4 8 mM Na2HPO4<br />
2 mM KH2PO4<br />
3 mM KCl<br />
121
PCR-Lysepuffer 50 mM KCl<br />
Material <strong>und</strong> Methoden<br />
[60] 10 mM Tris, pH 8,3<br />
0,45 % Tween ® 20<br />
0,45 % Nonidet P-40<br />
SDS-Elektrodenpuffer 250 mM Tris<br />
1 x 19 mM Glyzin<br />
0,35 mM SDS<br />
SSC 3 M NaCl<br />
20 x 300 mM C6H5Na3O7 . 2H2O<br />
TAE 800 mM Tris<br />
20 x 40 mM EDTA<br />
TBS 10 mM Tris<br />
10 x; pH 7,4 100 mM NaCl<br />
TBST (TBS plus Tween ® 20) 10 mM Tris<br />
1 x, pH 7,4 100 mM NaCl<br />
0,1 % Tween ® 20<br />
TE-Puffer 10 mM Tris-HCl, pH 8,1<br />
1 mM EDTA<br />
Transferpuffer für Immunblots 48 mM Tris<br />
39 mM Glyzin<br />
20 % Methanol<br />
Weitere Lösungen <strong>und</strong> Puffer sind in den entsprechenden Abschnitten aufgeführt.<br />
1.3 Nährmedien<br />
1.3.1 Nährmedien für Bakterien<br />
Standard I-Medium 15 g/l Trypton<br />
5 g/l Hefeextrakt<br />
1 g/l Glukose<br />
6 g/l NaCl<br />
122
Material <strong>und</strong> Methoden<br />
Das Medium wurde als Fertigpulver geliefert (Merck, Darmstadt). Zur Herstellung des<br />
Flüssigmediums wurden 25 g Pulver in 1 l ddH2O gelöst <strong>und</strong> autoklaviert. Zur Herstellung von<br />
Agarplatten wurden dem Medium vor dem Autoklavieren 1,5 % Bacto-Agar (Difco, USA)<br />
zugesetzt. Zur Herstellung von Selektionsmedium oder –Platten wurden dem autoklavierten,<br />
abgekühlten Medium 30 µg/ml Kanamycin oder 100 µg/ml Ampicillin zugesetzt.<br />
1.3.2 Medien für eukaryotische Zellen<br />
Alle Substanzen für die Zellkultur stammten von Gibco BRL (USA), Sigma (Mannheim) oder<br />
Biochrom (Berlin). Die flüssigen Nährmedien wurden vor der Verwendung wie folgt<br />
supplementiert:<br />
DMEM 10 % Hitzeinaktiviertes FCS<br />
für LMH <strong>und</strong> D2 1 mM Natriumpyruvat<br />
2 mM Glutamin<br />
100 U/ml Penicillin<br />
100 µg/ml Streptomycin<br />
Williams’ Medium E 1,5 % DMSO<br />
für PDHs 1 nM Insulin<br />
10 µM Hydrokortison<br />
15 mM HEPES, pH 7,2<br />
100 U/ml Penicillin<br />
100 µg/ml Streptomycin<br />
Trypsin-Lösung 0,25 % Trypsin<br />
1 mM EDTA-4Na<br />
Alle eukaryotischen Zellen wurden unter einer Wasserdampf-gesättigten 5 % CO2-Luft-<br />
Atmosphäre <strong>und</strong> bei 37°C kultiviert.<br />
1.4 Biologisches Material<br />
1.4.1 Bakterienstämme<br />
XL1 blue (Stratagene) wurde für die Amplifikation von Plasmiden verwendet,<br />
Genotyp: recA1, endA1, gyrA96, thi-1, hsdR17,<br />
supE44, relA1, lac [F'proAB lacI q Z?M15 Tn10 (Tet r )]<br />
123
1.4.2 Eukaryotische Zellen<br />
LMH Hepatomzelllinie aus Hühnern<br />
Material <strong>und</strong> Methoden<br />
D2 Hepatomzelllinie aus Hühnern (LMH), die stabil mit dem DHBV-Genom<br />
transfiziert ist (fre<strong>und</strong>licherweise zur Verfügung gestellt von J. Summers <strong>und</strong> W.<br />
Mason, USA)<br />
PDHs primäre Entenhepatozyten aus Entenembryonen, primary duck hepatocytes<br />
1.4.3 Viren<br />
Enten-Hepatitis B-Virus Als Inokulum diente DHBV-haltiges Gänseserum, das ungefähr<br />
1.5 Antikörper<br />
1.5.1 Primärantikörper <strong>und</strong> Antiseren<br />
1,1 x 10 10 Genomäquivalente pro ml enthielt, wie durch eine Dot<br />
Blot-Analyse von viralen Partikeln in Fraktionen nach CsCl-<br />
Gradientenzentrifugation festgestellt wurde. Die ungefähre Zahl<br />
der SVPs wurde mittels eines kalibrierten Immunblots bestimmt,<br />
bei dem gereinigtes, rekombinantes preS-Peptid<br />
(fre<strong>und</strong>licherweise zur Verfügung gestellt von S. Urban,<br />
Universität Heidelberg) als Standard diente. Die Berechnungen<br />
basierten auf der Annahme, dass von den ca. 100 viralen<br />
Oberflächenproteinen ungefähr 20 L-Proteine sind [25, 50] <strong>und</strong><br />
ergaben für das verwendete Serum ungefähr 10 13 SVPs/ml.<br />
Bezeichnung Spezies Antigen Herkunft/Referenz<br />
anti-PreS-KpnI Kaninchen PreS-Teil des L (AS 44-<br />
185) von DHBV<br />
anti-PreS-1-41 Kaninchen PreS-Teil des L (AS 1-<br />
41) von DHBV<br />
anti-PreS-DPSI Kaninchen PreS-Teil des L (AS 1-<br />
131) von DHBV<br />
[184]<br />
S. Urban, Heidelberg<br />
[191]<br />
anti-Core D103 Kaninchen Core-Protein H. Schaller, Heidelberg<br />
anti-Aktin Maus Aktin Sigma, Deisenhofen<br />
anti-GFP Kaninchen GFP Santa Cruz Biotechnology,<br />
USA<br />
anti-Caveolin-1 Kaninchen Caveolin Abcam, UK<br />
124
Material <strong>und</strong> Methoden<br />
Bezeichnung Spezies Antigen Herkunft/Referenz<br />
anti-Caveolin-1 Kaninchen Caveolin Transduction Laboratories,<br />
1.5.2 Sek<strong>und</strong>ärantikörper<br />
Bezeichnung Spezies Konjugat Herkunft<br />
anti-Maus-HRPO Ziege HRPO Dianova, Hamburg<br />
anti-Kaninchen-HRPO Ziege HRPO Dianova, Hamburg<br />
anti-Maus-488 Ziege Alexa 488 Molecular Probes, Niederlande<br />
anti- Kaninchen-488 Ziege Alexa 488 Molecular Probes, Niederlande<br />
anti-Maus-594 Ziege Alexa 594 Molecular Probes, Niederlande<br />
anti- Kaninchen-594 Ziege Alexa 594 Molecular Probes, Niederlande<br />
anti-Maus IgG Kaninchen Ohne Sigma, Deisenhofen<br />
1.6 Molekularbiologische Materialien<br />
1.6.1 Synthetische Oligonukleotide<br />
Primer 1: 5’-GCG CTT TCC AAG ATA CTG GAG CCC AA-3’<br />
Primer 2: 5’-CTG GAT GGG CCG TCA GCA GGA TTA TA -3’<br />
Primer 3: 5’-CCC TGT GTA GTC TGC CAG AAG TCT TC -3’<br />
Die Sequenzen der Primer stammen aus [60]. Sie wurden von Invitrogen bezogen.<br />
1.6.2 Plasmide<br />
Plasmidname Inhalt Referenz/Herkunft<br />
DHBV16t-27 Tandem-Sequenz des DHBV-<br />
Genoms 16<br />
DHBV wt 1,2-faches Genom des DHBV,<br />
pDHBV16/1.1<br />
DHBV D1 Wie oben, Punktmutationen in<br />
preS -Region (siehe Kapitel III.4)<br />
DHBV D2 Wie oben, Punktmutationen in<br />
preS -Region (siehe Kapitel III.4)<br />
USA<br />
Kaninchenserum Kaninchen HIPK2 H. Sirma, Hamburg<br />
[196]<br />
[51]<br />
E. Hildt, Berlin<br />
E. Hildt, Berlin<br />
125
Material <strong>und</strong> Methoden<br />
Plasmidname Inhalt Referenz/Herkunft<br />
DHBV D1/2 Wie oben, Punktmutationen in<br />
1.6.3 Enzyme <strong>und</strong> Kits<br />
Enzym Herkunft<br />
Proteinase K (PCR grade) Roche Diagnostics, Mannheim<br />
Plasmid Safe-DNase Epicentre, UK<br />
Taq-Polymerase Invitrogen, USA<br />
Kollagenase Roche, Mannheim<br />
Zur Herstellung radioaktiver Sonden wurde das ‚Readyprime II Random Prime Labeling System’<br />
von Amersham Pharmacia, Heidelberg, verwendet.<br />
Für die Präparation von Plasmiden wurde das ‚Plasmid Maxi Kit’ von Qiagen, Hilden,<br />
eingesetzt. Die Isolierung viraler DNA aus subzellulären Fraktionen erfolgte mit dem ‚High Pure<br />
Viral Nucleic Acid Kit’ von Roche, Mannheim.<br />
Für die Chemilumineszenz-Detektion wurde das ‚ECL Western Blotting Detection<br />
System’ von Pierce, USA, verwendet.<br />
1.6.4 Verwendete Substanzen <strong>und</strong> Inhibitoren<br />
Name Arbeitskonzentration Lösungsmittel Herkunft<br />
Calpaininhibitor I 10 µM DMSO Sigma<br />
Cholera Toxin Unter-<br />
einheit B (CTB)<br />
preS -Region (siehe Kapitel III.4)<br />
E. Hildt, Berlin<br />
Tubulin-GFP Tubulin mit GFP fusioniert Clontech, USA<br />
4 µg/ml H2O Sigma<br />
Cytochalasin D 20 µM DMSO Sigma<br />
Dichlorisocoumarin 10 µM DMSO Sigma<br />
Filipin III 10 µg/ml H2O Sigma<br />
FITC-CTB 4 µg/ml H2O Sigma<br />
Fluoreszein-Diazetat ca. 1 µg/ml DMSO Sigma<br />
preS -Peptid (AS 1-165) 0,47 µg/ml H2O S. Urban<br />
Methyl-ß-Cyclodextrin 10 mM H2O Sigma<br />
Mevinolin 10 µM DMSO Sigma<br />
MG132 10 µM DMSO Sigma<br />
Nocodazol 16,5 µM DMSO Sigma<br />
Nystatin 100 µg/ml H2O Calbiochem<br />
Paclitaxel 10 µM DMSO Molecular Probes<br />
126
Material <strong>und</strong> Methoden<br />
Name Arbeitskonzentration Lösungsmittel Herkunft<br />
Suramin 100 µg/ml DMSO Sigma<br />
TRITC-Phalloidin 4 µg/ml H2O Molecular Probes<br />
Wasserlösliches<br />
Cholesterin<br />
Die Arbeitskonzentrationen wurden durch Eskalationsstudien ermittelt <strong>und</strong> der Literatur<br />
entnommen. Alle Substanzen außer CTB (4°C) wurden bei -20°C gelagert.<br />
1.7 Geräte<br />
5 µg/ml H2O Sigma<br />
Bild-Scanner UMAX, Taiwan<br />
Filmentwickler Agfa Curix 60 PMA Bode, Hamburg<br />
Fluoreszenzmikroskop Zeiss, Jena<br />
Fluor-S MultiImager Bio-Rad, USA<br />
Fraction Recovery System Beckman, USA<br />
Konfokales Laserscanningmikroskop LSM 510 Meta Zeiss, Jena<br />
Mini-Ultrazentrifuge Sorvall Discovery M120 Hitachi, Japan<br />
Mikroskop Olympus, Japan<br />
PhosphoImager Fujix Bas Raytest, Straubenhardt<br />
Photometer 3.000 pro Pharmacia, Freiburg<br />
Photosystem für Gelaufnahmen MWG, Ebersberg<br />
RoboCycler Gradient 40 Stratagene, Niederlande<br />
SDS-PAGE-Apparatur Amersham, Freiburg<br />
Semi-Dry-System Trans-Blot SD Bio-Rad, USA<br />
Tischzentrifuge Centrifuge 5415 C Eppendorf, Hamburg<br />
Tischzentrifuge Centrifuge 5415 R Eppendorf, Hamburg<br />
Ultrazentrifuge Optima LE-80K Beckman, USA<br />
UV-Stratalinker Stratagene, Niederlande<br />
127
2. Methoden<br />
2.1 Zellkultur von Zelllinien <strong>und</strong> PDHs<br />
Material <strong>und</strong> Methoden<br />
Alle Arbeiten in Zusammenhang mit der Zellkultur erfolgten in einer Sterilbank zur Vermeidung<br />
bakterieller oder sonstiger Kontaminationen. Die verwendeten Medien <strong>und</strong> Chemikalien wurden<br />
mit sterilen Lösungsmitteln angesetzt, sterilfiltriert oder autoklaviert. Alle Plastikwaren, die in der<br />
Zellkultur verwendet wurden, stammten von Falcon, USA.<br />
Zum Passagieren von nahezu konfluent gewachsenen LMH- oder D2-Zellen in 250 ml<br />
Kulturflaschen wurde das Medium vollständig abgesaugt <strong>und</strong> die Zellen mit 0,25 % Trypsin-<br />
EDTA-Lösung überschichtet. Die Lösung wurde durch Schwenken verteilt <strong>und</strong> wieder<br />
abgesaugt. Dann folgte eine Inkubation für 2 min bei 37°C. Das Ablösen der Zellen wurde unter<br />
dem Mikroskop kontrolliert <strong>und</strong> die Zellen durch Klopfen von der Kulturflasche gelöst. Der<br />
Zellrasen wurde anschließend vorsichtig mit Medium abgespült <strong>und</strong> ein Zehntel der Suspension<br />
in eine neue Kulturflasche zur weiteren Kultivierung gegeben. Primäre Entenhepatozyten<br />
wurden nach der ersten Ausplattierung nicht passagiert.<br />
2.1.1 Transfektion<br />
Um Mikrotubuli in primären Entenhepatozyten sichtbar zu machen, wurden die Zellen zwei<br />
Tage, nachdem sie auf Glasplättchen ausgesät wurden, mit 4 µg Plasmid transfiziert. Dieses<br />
Plasmid kodierte für das Fusionsprotein Tubulin-GFP (Clontech, USA) <strong>und</strong> machte so die<br />
<strong>Untersuchung</strong> der Mikrotubuli ohne die Anwendung einer indirekten Immunfluoreszenzfärbung<br />
möglich. Die Transfektion erfolgte mit Hilfe von FuGENE 6 (Roche, Mannheim) nach den<br />
Angaben des Herstellers. 80 µl Kulturmedium ohne Zusätze <strong>und</strong> 20 µl FuGENE 6 wurden<br />
vermischt, bevor 4 µg der Plasmid-DNA zugegeben wurden. Nach einer Inkubation für 5 min bei<br />
Raumtemperatur wurde der Ansatz auf die Zellen gegeben. Einen Tag nach der Transfektion<br />
wurde das Medium gewechselt <strong>und</strong> die Transfektionseffizienz mit einem Lebendzellmikroskop<br />
bestimmt. Diese lag bei ca. 1 % der Zellen. Anschließend wurden die Zellen mit Nocodazol<br />
behandelt, um den Effekt der Substanz auf die Mikrotubuli untersuchen zu können.<br />
2.1.2 Produktion <strong>und</strong> Ernte rekombinanter Hepadnaviren<br />
Rekombinante Hepadnaviren können nach Transfektion von wt-DHBV- oder von mutierten<br />
DHBV-Genomen in LMH-Zellen aus dem Zellkulturüberstand gewonnen werden. In der<br />
Hühnerkarzinomzelllinie laufen alle späten Schritte einer Infektion so ab, dass komplette Viren<br />
<strong>und</strong> subvirale Partikel assembliert <strong>und</strong> anschließend auch sekretiert werden.<br />
128
Material <strong>und</strong> Methoden<br />
LMH-Zellen wurden nicht-konfluent in einer 6-Vertiefungsplatte ausplattiert. Am<br />
nächsten Tag wurde das Medium gewechselt <strong>und</strong> die Zellen transfiziert. Pro Platte wurde ein<br />
Konstrukt verwendet. Es wurden pro Vertiefung 2 µg Plasmid-DNA mit Hilfe von FuGENE 6<br />
nach den Angaben des Herstellers transfiziert. Am nächsten Tag wurde das Medium<br />
gewechselt. Etwa drei Tage nach der Transfektion wurde begonnen, den Kulturüberstand der<br />
transfizierten Zellen zu sammeln, ab diesem Zeitpunkt sollten die rekombinanten Viren<br />
sekretiert werden. Der Überstand wurde über drei bis vier Tage gesammelt. Anschließend<br />
wurden die Zellen für die Analyse der viralen DNA (replikative Intermediate) geerntet <strong>und</strong> die<br />
Viren im Zellkulturüberstand mit PEG gefällt (siehe IV.2.9.1).<br />
2.2 Anlegen einer Kultur primärer Entenhepatozyten<br />
Hepadnaviren sind nicht in der Lage, Zelllinien zu infizieren. Wichtige Wirtsfaktoren, die für die<br />
Etablierung einer produktiven Infektion essentiell sind, scheinen in diesen Zellen zu fehlen. Um<br />
Infektionsversuche durchführen zu können, mussten primäre Hepatozyten als Wirtszellen für<br />
das Enten-Hepatitis B-Virus präpariert werden.<br />
2.2.1 Präparation von primären Entenhepatozyten<br />
Um Entenhepatozyten für Infektionsversuche zu isolieren, wurden Enteneier 21 Tage bei 37°C<br />
<strong>und</strong> wasserdampfgesättigter Atmosphäre in einem Brutschrank inkubiert. Anschließend wurden<br />
die embryonierten Eier geöffnet, die Leber der Embryonen entfernt <strong>und</strong> in kleine Stücke<br />
geschnitten. Die Homogenate wurden dann in je 3 ml einer sterilen Kollagenase-Lösung (0,5 %<br />
Kollagenase in Williams’ Medium E) gegeben <strong>und</strong> für 45 min bei 37°C im Wasserbad inkubiert,<br />
um den Zellverband zu zerstören <strong>und</strong> die Zellen zu vereinzeln. Dabei wurden die Röhrchen in<br />
Abständen von 5 min geschüttelt. Danach wurden die Zellen bei 800 rpm für 4 min<br />
abzentrifugiert, der Überstand entfernt <strong>und</strong> das Pellet in 5 ml Williams’ Medium E ohne Zusätze<br />
resuspendiert, um die Zellen von Verunreinigungen zu befreien. Die Waschschritte wurden<br />
insgesamt dreimal wiederholt. Das resultierende Zellpellet wurde in Wachstumsmedium (siehe<br />
IV.1.4.2) aufgenommen <strong>und</strong> normalerweise auf 12-Vertiefungs -Platten mit einer Zelldichte von<br />
ca. 5 x 10 5 Hepatozyten pro Vertiefung ausplattiert. Dabei wurde darauf geachtet, dass DHBV-<br />
positive <strong>und</strong> -negative Lebern (siehe IV.2.2.2) getrennt wurden. Nachdem sich die Zellen<br />
abgesetzt hatten (ca. 4 h nach Ausplattierung), wurde das Medium gewechselt. Typischerweise<br />
wurden für folgende Versuche Kulturen verwendet, die drei bis fünf Tage alt waren, <strong>und</strong> jeden<br />
Tag das Medium gewechselt.<br />
Die Primärkulturen, die für Infektionsexperimente verwendet wurden, waren<br />
Mischkulturen, in denen die Hepatozyten neben anderen Zellen der Leber (sog. Nicht-<br />
Parenchymzellen) vorkamen. Bis zu 30 % der Leberzellkultur bestand aus Nicht-Parenchym-<br />
zellen.<br />
129
2.2.2 Protein-Dot Blot zum Nachweis DHBV-infizierter Lebern<br />
Material <strong>und</strong> Methoden<br />
Vor Beginn der Experimente mit den primären Entenhepatozyten wurde überprüft, ob eine<br />
DHBV-Infektion der verwendeten embryonalen Lebern vorlag. Dies wurde mit Hilfe des Protein<br />
Dot-Blot getestet, diese Art des Immunblot verzichtet auf die Auftrennung der Proteine in einer<br />
SDS-PAGE.<br />
Serum der verwendeten Entenembryonen wurde in PBS seriell verdünnt <strong>und</strong> mit Hilfe<br />
einer Dot Blot-Apparatur auf eine Nitrozellulosemembran aufgebracht. Die Apparatur wurde<br />
folgendermaßen aufgebaut: Zuerst wurde ein in 1 x PBS angefeuchtetes 3 MM Whatmanpapier<br />
aufgelegt, dann folgte die angefeuchtete Membran. Die Dot Blot-Apparatur wurde<br />
zusammengesetzt <strong>und</strong> ein Unterdruck in der Auffangkammer erzeugt. Die Dots wurden einmal<br />
mit PBS gewaschen, anschließend die Proben (50 µl Volumen) aufgedottet <strong>und</strong> wieder<br />
gewaschen. Als Positivkontrolle wurde immer ein bekanntes, virämisches Serum verwendet.<br />
Die Membran wurde aus der Apparatur genommen <strong>und</strong> getrocknet. Dann wurde kurz mit<br />
Blocking-Reagenz inkubiert <strong>und</strong> sofort danach der Primärantikörper PreS-KpnI zugegeben.<br />
Dieser Antikörper war 1:20.000 in Blocking-Reagenz verdünnt. Nach 30 min Inkubation bei<br />
Raumtemperatur wurde die Membran kurz mit TBST gewaschen <strong>und</strong> anschließend mit dem<br />
Sek<strong>und</strong>ärantikörper (Verdünnung 1:20.000, anti-Kaninchen) 30 min inkubiert. Die Membran<br />
wurde dann dreimal mit TBST gewaschen <strong>und</strong> das Signal mit dem ECL-System (Pierce, USA)<br />
auf einem Röntgenfilm sichtbar gemacht.<br />
2.3 DHBV-Bindung, Internalisierung <strong>und</strong> produktive Infektion<br />
2.3.1 Bindungs- <strong>und</strong> Interferenzassay<br />
Um den Effekt von Suramin auf die Bindung der Viren zu untersuchen, die an die Zelloberfläche<br />
der PDH-Kulturen binden, wurden die Zellen entweder für 1 h mit 100 µg/ml Suramin<br />
vorbehandelt oder nicht behandelt. Anschließend wurden sie mit virämischem Serum inokuliert,<br />
so dass 220 GE pro Zelle erreicht wurden. Danach wurden die Zellen für 2 h bei 4°C inkubiert,<br />
um die virale Bindung, jedoch nicht die Aufnahme zu erlauben. Nach gründlichem Waschen<br />
wurden die Zellen in PCR- oder Lämmli-Lysepuffer geerntet.<br />
Um den Effekt neutralisierender Antikörper auf die virale Bindung zu untersuchen,<br />
wurde das Inokulum mit 10 µl der Antiseren DPSI, KpnI, Kaninchen-Kontrollserum oder 20 µg<br />
gereinigtes IgG für 30 min bei 37°C inkubiert <strong>und</strong> dann auf die Zellen gegeben. Die Kulturen<br />
wurden 2 h bei 4°C inkubiert, dann mit PBS gewaschen <strong>und</strong> in 200 µl PCR- oder Lämmli-<br />
Lysepuffer geerntet.<br />
Für die Analyse der GE-Abhängigkeit <strong>und</strong> Sättigung der viralen Bindung, wurden PDHs<br />
mit verschiedenen Mengen virämischen Serums wie oben beschrieben inkubiert <strong>und</strong> geerntet<br />
oder die Inkubationszeit bei 4°C wurde verändert.<br />
130
Material <strong>und</strong> Methoden<br />
Um zu untersuchen, ob DHBV an lipid rafts bindet, wurden die Zellen zuerst mit kaltem<br />
Medium gewaschen. Dann wurde virämisches Serum zugegeben <strong>und</strong> die Zellen mit dem Virus<br />
für 2 h bei 4°C inkubiert. Anschließend wurden die Kulturen extensiv mit eiskaltem PBS<br />
gewaschen <strong>und</strong> entweder direkt geerntet oder für verschiedenen Zeitspannen bei 37°C<br />
inkubiert <strong>und</strong> dann geerntet. Für die Ernte wurden die Zellen mit eiskaltem PBS gewaschen <strong>und</strong><br />
die Zellen in 1 ml eiskaltem TNE -X-Puffer (siehe IV.2.6.2) aus der Vertiefung geschabt.<br />
Anschließend wurden Detergenz-unlösliche Membrandomänen isoliert (siehe IV.2.6.2).<br />
2.3.2 Internalisierungs- <strong>und</strong> Protease-Schutzassay<br />
Um die Internalisierung viraler Partikel untersuchen zu können, wurde ein<br />
Internalisierungsassay etabliert. Nach der oben beschriebenen Bindung der viralen Partikel an<br />
die Zelloberfläche, wurden die Zellen gewaschen, um das Inokulum zu entfernen. Dann wurde<br />
neues Medium zugegeben <strong>und</strong> die Kulturen für verschiedene Zeitspannen bei 37°C inkubiert. In<br />
dieser Zeit sollte das an die Zelloberfläche geb<strong>und</strong>ene Virus in das Zellinnere aufgenommen<br />
werden. Zu unterschiedlichen Zeitpunkten wurde das Medium abgenommen, die Zellen<br />
gewaschen <strong>und</strong> 1 ml 0,25 % Trypsinlösung (Gibco BRL, USA) zugegeben. Die Kulturen wurden<br />
5 min bei 37°C inkubiert, anschließend in ein Eppendorf-Röhrchen überführt <strong>und</strong> durch<br />
Zentrifugation pelletiert. Das Pellet wurde entweder in 200 µl PCR- oder Lämmli-Lysepuffer<br />
lysiert.<br />
Sollte die Lokalisierung der viralen Partikel nach der Aufnahme untersucht werden,<br />
wurde das Pellet nach der oben beschriebenen Ernte mit PBS gewaschen, in 500 µl<br />
Homogenisierungspuffer aufgenommen <strong>und</strong> eine subzelluläre Fraktionierung durchgeführt<br />
(siehe IV.2.6.1).<br />
2.3.3 Infektion<br />
Für die Infektion der Hepatozyten mit DHBV wurde ein virämisches Gänseserum eingesetzt,<br />
das ca. 1,1 x 10 10 Genomäquivalente (GE) pro ml enthielt. Normalerweise wurden für Infek-<br />
tionen 220 GE pro Hepatozyte eingesetzt, es wurde über Nacht bei 37°C inkubiert <strong>und</strong><br />
anschließend das Medium gewechselt. Drei Tage nach der Inokulation wurde die produktive<br />
Infektion in den Kulturen getestet.<br />
2.3.4 pH-Schock<br />
Um extrazellulär geb<strong>und</strong>ene Viren nach einer Inokulation zu inaktivieren, wurde ein<br />
zweiminütiger pH-Schock mit einem Glyzin-Puffer pH 2,2 durchgeführt [60].<br />
131
Glyzin-Puffer: 50 mM Glyzin<br />
pH 2,2 150 mM Natriumchlorid<br />
2.4 Behandlungen<br />
2.4.1 Infektion der PDHs in Anwesenheit verschiedener Substanzen<br />
Material <strong>und</strong> Methoden<br />
Typischerweise wurde folgendes Behandlungsschema verfolgt: Das Medium in den Kulturen<br />
wurden gewechselt, anschließend wurde neues Medium, das eine inhibitorische Konzentration<br />
der Substanz enthielt, auf die Zellen gegeben <strong>und</strong> die Zellen für 1 h vorbehandelt. Danach<br />
wurde das Virus zugegeben <strong>und</strong> über Nacht zusammen mit der Substanz inkubiert. Dann<br />
wurden sowohl Virus als auch Substanz entfernt <strong>und</strong> die Zellen weitere drei Tage bis zur Ernte<br />
inkubiert.<br />
Um den Effekt neutralisierender Antikörper oder Suramin auf die primäre Infektion zu<br />
untersuchen, wurden virämisches Serum oder die Zellen mit den Substanzen wie in IV.2.3.5.<br />
beschrieben vorinkubiert (Vorbehandlung). Alternativ wurden 100 µg/ml Suramin oder 10 µl<br />
DPSI zusammen mit dem Inokulum (synchrone Behandlung) oder 2 h nach der Inokulation<br />
zugegeben (Nachbehandlung). Anschließend wurden die Zellen über Nacht bei 37°C inkubiert<br />
<strong>und</strong> ungeb<strong>und</strong>ene virale Partikel durch Waschen entfernt. Geb<strong>und</strong>ene, aber nicht internalisierte<br />
Viren wurden durch einen pH-Schock inaktiviert. Anschließend wurden die Zellen für weitere<br />
drei Tage inkubiert, bevor sie geerntet <strong>und</strong> durch Immunfärbung <strong>und</strong> -Blot analysiert wurden.<br />
2.4.2 Inhibition der Sek<strong>und</strong>ärinfektion<br />
PDHs wurden mit 220 GE pro Zelle über Nacht infiziert, dann wurde das Medium gewechselt<br />
<strong>und</strong> die Zellen weitere drei Tage inkubiert. Anschließend wurden auf die Kulturen entweder<br />
100 µg/ml Suramin oder 10 µl DPSI-Antiserum gegeben. Zu diesem Zeitpunkt beginnen die<br />
infizierten Zellen, Nachkommensviren zu sekretieren. Das Medium wurde täglich gewechselt<br />
<strong>und</strong> frische Substanzen wurden zugegeben. Nach weiteren drei Tagen wurden die Zellen fixiert<br />
<strong>und</strong> nach einer indirekten Immunfluoreszenzfärbung durch Mikroskopie analysiert.<br />
2.4.3 wash out-Assay<br />
Dieser Assay wurde verwendet, um die mögliche Reversibilität einer Behandlung zu<br />
untersuchen. Dazu wurden Viren für 2 h bei 4°C an die Zelloberfläche geb<strong>und</strong>en. Anschließend<br />
wurden die Zellen mit PBS gewaschen, um ungeb<strong>und</strong>enes Virus zu entfernen. Neues Medium,<br />
das die gewünschte Substanz enthielt, wurde zugegeben <strong>und</strong> die Platte bei 37°C inkubiert, um<br />
die vi rale Aufnahme zu gewährleisten. Zu bestimmten Zeitpunkten nach Beginn der Inkubation<br />
132
Material <strong>und</strong> Methoden<br />
wurden die Zellen gewaschen, um die Substanz zu entfernen. Nach weiteren drei Tagen<br />
wurden die Zellen auf die Etablierung einer produktiven Infektion untersucht. Dasselbe<br />
Vorgehen wurde auch bei unbehandelten Kontrollzellen angewandt.<br />
2.4.4 add in-Assay<br />
Mit diesem Assay sollte untersucht werden, zu welchem Zeitpunkt der Etablierung einer<br />
Infektion eine Behandlung mit inhibierenden Substanzen effizient ist. Viren wurden für 2 h bei<br />
4°C an die Zelloberfläche geb<strong>und</strong>en, dann wurde das Medium gewechselt <strong>und</strong> die Kulturen bei<br />
37°C weiter inkubiert. Zu unterschiedlichen Zeitpunkten wurde dann die verwendete Substanz<br />
zugegeben. Die Zellen wurden über Nacht weiter inkubiert, dann das Medium gewechselt <strong>und</strong><br />
dadurch die Substanz entfernt. Nach weiteren drei Tagen wurden die Kulturen auf eine<br />
produktive Infektion untersucht.<br />
2.4.5 Effekt verschiedener Substanzen auf eine etablierte Infektion<br />
Um den Effekt verschiedener Substanzen auf die virale Replikation <strong>und</strong> Sekretion zu<br />
untersuchen, wurden entweder Kulturen von D2-Zellen oder chronisch infizierte PDHs<br />
verwendet. Diese Kulturen wurden kontinuierlich zwischen 24 h <strong>und</strong> 72 h mit derselben Menge<br />
Substanz inkubiert, die auch für die Infektionsversuche eingesetzt wurde. Dann wurden sowohl<br />
die Zellen als auch die Zellkulturüberstände geerntet <strong>und</strong> auf virale Marker analysiert.<br />
2.4.6 Effekt der Substanzen auf die Lebensfähigkeit der Zellen<br />
Verschiedene Substanzen könnten sich nach längerer Inkubation toxisch auf die Zellen<br />
auswirken. Um dies zu überprüfen, wurden Kulturen für 24 h mit den jeweiligen Substanzen<br />
behandelt. Anschließend wurden die Zellen mit PBS gewaschen <strong>und</strong> mit 0,4 % Trypanblau<br />
(Gibco BRL, USA) in PBS für 3 min inkubiert. Nach einem weiteren Waschschritt wurden die<br />
Kulturen auf blau gefärbte Zellen untersucht <strong>und</strong> fotografiert. Die Aufnahme des Farbstoffes in<br />
die Zellen kommt nur zustande, wenn die Zellmembranen nicht mehr intakt sind <strong>und</strong> die<br />
Substanz in die Zellen durch Diffusion eindringt. Dunkel erscheinen daher Zellen, die<br />
geschädigt sind.<br />
Parallelkulturen wurden mit 10 µl Fluoreszein-Diazetat (eine Spatelspitze gelöst in 1 ml<br />
DMSO) für 5 min bei 37°C inkubiert. Anschließend wurde das Medium gewechselt <strong>und</strong> die<br />
Kultur unter dem Fluoreszenzmikroskop untersucht <strong>und</strong> Bilder gemacht. Nur lebende<br />
Hepatozyten nehmen das Fluoreszein-Diazetat auf <strong>und</strong> besitzen die entsprechende Lipase um<br />
es in den Fluoreszenzfarbstoff Fluoreszein zu überführen. Diese Zellen erscheinen im<br />
Fluoreszenzmikroskop dann grün.<br />
133
2.4.7 Behandlung von DHBV<br />
Material <strong>und</strong> Methoden<br />
Bei einigen Substanzen bestand die Möglichkeit, dass sie nicht auf die Zellen, sondern auf die<br />
viralen Partikel wirken. Um dies zu überprüfen, wurden 100 µl virämisches Serum für 1 h bei<br />
37°C in Williams’ Medium E mit der Substanz inkubiert. Serum ohne Substanz wurde als<br />
Kontrolle identisch behandelt. Dann wurden die viralen Partikel entweder mit PEG gefällt oder<br />
pelletiert (siehe IV.2.9.1). Das Pellet wurde in PBS aufgenommen <strong>und</strong> damit Infektionsstudien<br />
durchgeführt oder die physikalischen Eigenschaften (siehe IV.2.9.2) untersucht.<br />
2.5 Nachweis viraler DNA durch Polymerase-Kettenreaktion (PCR)<br />
Die Polymerase-Kettenreaktion (polymerase chain reaction, PCR) ist eine Methode zur in vitro<br />
Vermehrung (Amplifikation) definierter Nukleinsäureabschnitte mit Hilfe des Enzyms DNA-<br />
Polymerase [197]. Sie erlaubt den schnellen <strong>und</strong> sensitiven Direktnachweis kleinster Mengen<br />
von DNA.<br />
In dieser Arbeit wurde die PCR dazu verwendet, virale DNA zu amplifizieren <strong>und</strong><br />
anschließend im Agarosegel semiquantitativ nachzuweisen. Für den Nachweis von rcDNA<br />
wurde das Primerpaar P1 <strong>und</strong> P2 verwendet, für den Nachweis der cccDNA das Primerpaar P1<br />
<strong>und</strong> P3.<br />
2.5.1 Nachweis der viralen rcDNA in Zelllysaten<br />
Die virale Bindung <strong>und</strong> Aufnahme in PDHs wurde durch eine semiquantitative PCR untersucht.<br />
Der Zellrasen einer Vertiefung wurde nach einem erfolgten Versuch gründlich mit 1 x PBS<br />
gewaschen, dann wurden 200 µl PCR-Lysepuffer in die Vertiefung gegeben <strong>und</strong> 5 min bei 37°C<br />
inkubiert. Der Zellrasen wurde in ein Reaktionsgefäß überführt <strong>und</strong> weitere 5 min bei RT<br />
inkubiert. Anschließend wurden die in der Lösung befindlichen Proteine mit Proteinase K<br />
verdaut. Dazu wurde das Lysat mit 0,1 mg/ml Enzym versetzt <strong>und</strong> für 2 h bei 56°C inkubiert.<br />
Dann wurde das Enzym durch eine Inkubation bei 95°C für 10 min inaktiviert <strong>und</strong> 5 µl des<br />
Lysats für die PCR eingesetzt. Es wurde folgender Reaktionsansatz für die PCR verwendet:<br />
5 µl DNA<br />
0,5 µl Primer P1 (50 pmol)<br />
0,5 µl Primer P2 (50 pmol)<br />
0,5 µl Taq-Polymerase (2,5 Units)<br />
0,5 µl dNTP -Mix (0,2 mM)<br />
5 µl 10 x PCR-Puffer<br />
1,5 µl MgCl2<br />
ad 50 µl ddH2O<br />
134
Material <strong>und</strong> Methoden<br />
Zur Vermeidung von Verdunstung flüssiger Bestandteile während der Reaktion wurden<br />
die Ansätze mit Mineralöl überschichtet.<br />
Als Kontrolle einer möglichen Kontamination wurde zu einem Ansatz keine DNA<br />
hinzugegeben, sondern nur ddH2O. Außerdem wurde als Positivkontrolle <strong>und</strong> Standard<br />
virämisches Serum mit bekanntem Virusgehalt verwendet, das in PCR-Lysepuffer verdünnt<br />
wurde.<br />
Das Temperaturprogramm für die Durchführung der PCR mit 25 Zyklen war wie folgt:<br />
1 min 94°C Denaturierung<br />
3 min 72°C DNA-Synthese<br />
Der Annealing-Schritt wurde ausgelassen, um eine Bindung der Amplifikate<br />
untereinander zu verhindern.<br />
10 µl der PCR-Produkte wurden anschließend auf ein 1 % Agarosegel, das<br />
Ethidiumbromid enthielt, aufgetragen <strong>und</strong> analysiert.<br />
2.5.2 Reinigung <strong>und</strong> Nachweis der viralen cccDNA in infizierten Zellen<br />
Um zu überprüfen, ob eine produktive Infektion in den mit DHBV inkubierten <strong>und</strong> behandelten<br />
Zellen stattgef<strong>und</strong>en hatte, wurde die im Zellkern produktiv infizierter Zellen amplifizierte virale<br />
cccDNA isoliert. Dies geschah nach einem modifizierten Protokoll von J. Summers [161].<br />
Der Zellrasen wurde 10 min bei 37°C mit Trypsin-Lösung inkubiert, um an die<br />
Oberfläche geb<strong>und</strong>enes Virus zu entfernen. Anschließend wurden die Zellen in der Lösung<br />
aufgenommen, in ein Eppendorfgefäß überführt <strong>und</strong> für 10 min bei 4°C <strong>und</strong> 13.000 rpm<br />
pelletiert. Dann wurde das Pellet in 1 ml SDS -Lysepuffer aufgenommen <strong>und</strong> für 1 h unter<br />
Schütteln bei 37°C inkubiert, um die virale DNA freizusetzen. Anschließend wurden 250 µl 2,5<br />
M KCl zugegeben, um Proteine <strong>und</strong> an sie geb<strong>und</strong>ene DNA zu fällen. Nach gründlichem<br />
Vortexen wurden die nun unlöslichen Bestandteile durch Zentrifugieren von den löslichen<br />
abgetrennt. Im Überstand war nun die virale cccDNA vorhanden, im erhaltenen Pellet die<br />
replikativen Intermediate. Der Überstand wurde mit 1 ml Phenol (Biomol, Hamburg) gründlich<br />
gemischt, um eventuell noch vorhandene Proteine zu entfernen. Nach einer kurzen<br />
Zentrifugation, die zur Trennung der organischen von der wässrigen Phase führte, wurde die<br />
wässrige Phase (enthält die DNA) abgenommen <strong>und</strong> mit 1 ml Chloroform gemischt, um Reste<br />
von Phenol zu entfernen. Nach Zentrifugation bei 13.000 rpm <strong>und</strong> 4°C für 5 min wurde die<br />
wässrige Phase wieder in ein neues Eppendorfgefäß überführt. Dazu wurden nun 2 ml eiskaltes<br />
Ethanol <strong>und</strong> 100 µl 3 M Natriumazetat gegeben, um die DNA zu fällen. Dieser Ansatz wurde für<br />
1 h bei -80°C inkubiert <strong>und</strong> anschließend für 30 min bei 13.000 rpm <strong>und</strong> bei 4°C zentrifugiert.<br />
Das erhaltene Pellet wurde in 10 µl H20 aufgenommen. Davon wurden entweder 5 µl direkt für<br />
die PCR eingesetzt oder ein DNA-Verdau mit Plasmid Safe-DNase durchgeführt (siehe<br />
IV.2.5.3).<br />
135
Material <strong>und</strong> Methoden<br />
Die PCR zum Nachweis der cccDNA wurde unter denselben Bedingungen wie in<br />
IV.2.5.1 beschrieben durchgeführt. Es wurde jedoch anstelle von Primer 2 der Primer 3<br />
verwendet, der an die Region der cccDNA bindet, die im Zellkern erst vervollständigt wird.<br />
Durch Verwendung dieser Primerkombination wird präferentiell cccDNA amplifiziert.<br />
2.5.3 Plasmid Safe DNAse-Verdau<br />
In einigen Fällen wurde die durch obige Prozedur erhaltene DNA mit einer DNase verdaut, die<br />
präferentiell einzelsträngige DNA verdaut. Diese Plasmid Safe DNase (Epicentre, UK) sollte die<br />
partiell einzelsträngige rcDNA <strong>und</strong> die replikativen Intermediate verdauen, während die cccDNA<br />
nicht verdaut werden sollte. Dieser Verdau wurde durchgeführt, um eventuell vorhandene<br />
Verunreinigungen, die in der PCR ein positives Signal geben könnten, auszuschließen.<br />
Die 10 µl der DNA-Lösung wurden zuerst 10 min bei 95°C inkubiert, um den Verdau<br />
effizienter zu machen. Dann wurde der Verdau angesetzt:<br />
10 µl DNA<br />
1,6 µl 10 x Reaktionspuffer<br />
1,2 µl ATP (25 mM)<br />
1 µl Plasmid Safe DNase<br />
2,2 µl H2O<br />
Dieser Ansatz wurde für 2 h bei 37°C inkubiert; anschließend wurde die DNase durch<br />
eine Inkubation von 30 min bei 70°C inaktiviert. 5 µl der Lösung wurden dann für eine PCR<br />
eingesetzt.<br />
2.6 Subzelluläre Fraktionierung<br />
2.6.1 Fraktionierung durch sequentielle Zentrifugation<br />
Um Zellen in ihre verschieden großen Bestandteile aufzutrennen, wurden sie durch sequentielle<br />
Zentrifugation fraktioniert [181]. Dazu wurden die Kulturen zweimal mit 1 x PBS gewaschen <strong>und</strong><br />
in 500 µl Homogenisierungspuffer abgeschabt. Die Zellen wurden dann in einem Zellpotter<br />
aufgeschlossen. Anschließend wurde das Ganze für 1 min bei 13.200 rpm in einer<br />
Tischzentrifuge bei 4°C zentrifugiert, um unaufgeschlossene Zellen <strong>und</strong> große Membranstücke<br />
abzutrennen. Der Überstand wurde in ein neues Reaktionsgefäß überführt, das Pellet in<br />
Lämmli- oder PCR-Lysepuffer aufgenommen. Der Überstand wurde dann für 10 min bei 50.000<br />
g zentrifugiert, um die Endosomen von den restlichen Zellbestandteilen zu trennen. Der<br />
Überstand wurde wieder in ein neues Reaktionsgefäß überführt. Er wurde für 18 min bei<br />
136
Material <strong>und</strong> Methoden<br />
430.000 g zentrifugiert. Das Pellet enthielt vorwiegend Mikrosomen <strong>und</strong> wurde daher als<br />
Mikrosomen-Fraktion bezeichnet. Der Überstand dieser letzen Zentrifugation wurde als Zytosol-<br />
Fraktion bezeichnet.<br />
Aus diesen Fraktionen wurde die virale DNA mit Hilfe des ‚High Pure Viral Nucleic Acid<br />
Kits’ (Roche, Mannheim) nach den Angaben des Herstellers isoliert.<br />
Homogenisierungspuffer: 50 mM Tris-HCl, pH 7,5<br />
5 mM MgCl2<br />
30 mM KCl<br />
5 mM DTT<br />
250 mM Sucrose<br />
1 Tablette Complete ® Protease-Inhibitor-Cocktail (Roche, Mannheim)<br />
2.6.2 Isolierung Detergenz-unlöslicher Membranen<br />
Detergenz-unlösliche Membranen (detergent insoluble membranes, lipid rafts, DIM) zeichnen<br />
sich im Vergleich zu der sie umgebenden Membran durch einen hohen Gehalt an Cholesterin<br />
<strong>und</strong> Sphingolipiden aus. Die Anhäufung dieser Lipide führt dazu, dass dieser Membranteil bei<br />
4°C nicht in einem Detergenz-haltigen Puffer löslich ist [198]. Diese Eigenschaft wurde genutzt,<br />
um DIMs aus PDHs zu isolieren <strong>und</strong> zu untersuchen, ob DHBV nach der Bindung in DIMs<br />
lokalisiert.<br />
Nach der Bindung viraler Partikel (440 MGE) oder rekombinantem preS-Peptid (final<br />
0,47 µg/ml, AS 1-165) an mit MßCD behandelte oder unbehandelte Kulturen wurden diese mit<br />
eiskaltem PBS gewaschen. Dann wurde 1 ml eiskaltes PBS in die Vertiefung der verwendeten<br />
6-Vertiefungsplatte gegeben <strong>und</strong> die Zellen darin abgeschabt. Die Zellen wurden dann durch<br />
eine Zentrifugation bei 1.000 g <strong>und</strong> bei 4°C für 5 min pelletiert. Das Pellet wurde in 500 µl TNE-<br />
X-Puffer aufgenommen <strong>und</strong> 30 min auf Eis inkubiert. In dieser Zeit lösten sich die Detergenz-<br />
löslichen Membranteile. Anschließend wurde der Ansatz bei 13.000 g <strong>und</strong> 4°C für 10 min<br />
zentrifugiert. Das Pellet enthielt nun die DIMs, der Überstand die DSMs (detergent soluble<br />
membranes, Detergenz-lösliche Membranen). Das Pellet wurde in 200 µl Lämmli-Lysepuffer<br />
aufgenommen, in den Überstand wurden 125 µl Lämmli-Lysepuffer gegeben. Für den<br />
Immunblot wurden jeweils 5 % eingesetzt.<br />
TNE-X-Puffer: 25 mM Tris-HCl, pH 7,4<br />
100 mM NaCl<br />
1 mM EDTA<br />
0,1 % Triton X-100<br />
1 Tablette Complete ® Protease-Inhibitor-Cocktail (Roche, Mannheim)<br />
137
2.7 Indirekte Immunfluoreszenzfärbung <strong>und</strong> Mikroskopie<br />
2.7.1 Indirekte Immunfluoreszenzfärbung<br />
Material <strong>und</strong> Methoden<br />
Die Kulturen wurden einmal mit PBS gewaschen <strong>und</strong> entweder mit Methanol/Azeton (1:1,<br />
eiskalt) oder 3,5 % Paraformaldehyd für 10 min bei RT fixiert. Die Zellen wurden anschließend<br />
mit PBS rehydriert <strong>und</strong> für 1 h mit dem entsprechenden Antikörper in der korrekten Verdünnung<br />
inkubiert. Dann wurden die Zellen dreimal mit PBS gewaschen <strong>und</strong> mit geeignetem<br />
Sek<strong>und</strong>ärantikörper für 1 h inkubiert, der mit einem Alexa-Farbstoff gekoppelt war. Die<br />
Zellkerne wurden mit Hoechst (finale Konzentration 4 µg/ml) gefärbt. Dann wurden die<br />
gefärbten Zellen entweder direkt in der Vertiefung untersucht oder die Glasplättchen wurden<br />
eingebettet.<br />
Zur Visualisierung der Aktinfasern wurden die Kulturen zweimal mit PBS gewaschen<br />
<strong>und</strong> dann für 10 min mit 3,7 % Paraformaldehyd fixiert. Nach einem Waschschritt mit PBS<br />
wurden die fixierten Zellen mit eiskaltem Azeton für 5 min behandelt <strong>und</strong> anschließend wieder<br />
gewaschen. Dann wurden die Glasplättchen mit 1 % BSA (bovines Serumalbumin, von Sigma)<br />
in PBS für 30 min geblockt. Anschließend wurde das TRITC-markierte Phalloidin (in PBS<br />
verdünnt, finale Konzentration 4 µg/ml) für 30 min auf die fixierten Zellen gegeben. Dieses<br />
bindet spezifisch F-Aktin. Nach gründlichem Waschen mit PBS wurden die Zellen kurz in 0,1 %<br />
Triton X-100 in PBS inkubiert. Dann wurden die Glasplättchen kurz gewaschen <strong>und</strong> mit Mowiol<br />
eingebettet. Konfokale Aufnahmen wurden mit einem Zeiss Laserscanningmikroskop gemacht.<br />
Um die Inaktivierung von lipid rafts zu kontrollieren, wurden Kulturen erst mit den<br />
verschiedenen inhibitorischen Substanzen behandelt <strong>und</strong> dann für 20 min mit 5 µg/ml FITC-<br />
CTB inkubiert. Konfokale Aufnahmen wurden mit einem Zeiss Laserscanningmikroskop<br />
gemacht.<br />
2.7.2 Konfokale Lasermikroskopie<br />
Primäre Entenhepatozyten wurden auf Glasplättchen gezogen, behandelt <strong>und</strong> fixiert. Die Bilder<br />
wurden mit einem konfokalen Laserscanningmikroskop aufgenommen (LSM 510 META, Zeiss,<br />
Germany) <strong>und</strong> mit der LSM5 Image Browser Software prozessiert.<br />
2.7.3 Elektronenmikroskopie<br />
Primäre Entenhepatozyten wurden in Kapillaren transferiert, um die Verarbeitung zu erleichtern.<br />
Diese wurden dann mit 2,5 % Glutaraldehyd in PBS für 20 min bei RT fixiert, dann gewaschen<br />
<strong>und</strong> für 30 min mit 1 % OsO4 in PBS post-fixiert. Für die ultradünnen Schnitte wurden die<br />
Proben graduell mit Ethanol dehydriert <strong>und</strong> in ERL-Harz eingebettet. Die Schnitte wurden mit<br />
138
Material <strong>und</strong> Methoden<br />
2 % Uranylazetat <strong>und</strong> Bleizitrat gefärbt. Die elektronenmikroskopischen Aufnahmen wurden mit<br />
einem Philips CM 120 Transmissions-Elektronenmikroskop bei 60 kV aufgenommen. Dies<br />
wurde dankenswerterweise in der Abteilung für Elektronenmikroskopie <strong>und</strong> Mikromanipulation<br />
am Heinrich-Pette-Institut von Dr. Hohenberg <strong>und</strong> seinen Mitarbeitern durchgeführt.<br />
2.8 Immunblot<br />
Nach vorangehender Behandlung, Transfektion oder Infektion wurden Lysate der<br />
eukaryotischen Zellen hergestellt, um diese weiter analysieren zu können. Der Zellrasen in der<br />
Vertiefung einer 12-Vertiefungsplatte wurde mit 1 x PBS mehrfach gewaschen. Dann wurde für<br />
den Immunblot eine entsprechende Menge Lämmli-Lysepuffer direkt in die Vertiefung der<br />
Zellkulturplatte gegeben (200 µl Puffer in eine Vertiefung einer 12-Vertiefungsplatte). Die Zellen<br />
wurden mit einer Pipettenspitze abgeschabt <strong>und</strong> in ein Reaktionsgefäß überführt.<br />
2.8.1 SDS-Polyacrylamid-Gelelektrophorese (SDS -PAGE) <strong>und</strong> Immunblot<br />
Alle Proben wurden bei 99°C für 5 min gekocht, anschließend wurde ein Aliquot (5 bis 10 %)<br />
jeder Probe durch eine denaturierende SDS-PAGE fraktioniert. Als Größenstandard wurden 3 µl<br />
‚Prestained Protein Marker’ von Invitrogen aufgetragen. Anschließend wurden die Proteine auf<br />
eine Nitrozellulose-Membran (Schleicher <strong>und</strong> Schüll, Dassel) transferiert. Nach Sättigen der<br />
Membran mit 3 % Trockenmilch (Biorad, USA) in TBS (Tris buffered saline), wurde sie für 1 h<br />
bei RT mit dem Erstantikörper in der korrekten Verdünnung inkubiert. Nach mehrfachem<br />
Waschen mit TBST (TBS plus 0,1 % Tween ® ) wurde die Membran mit dem Zweitantikörper für<br />
1 h inkubiert. Dieser war mit der Meerrettich-Peroxidase gekoppelt. Nach mehreren<br />
Waschgängen wurden die Proteine mittels indirekter Chemilumineszenz (Pierce, USA)<br />
visualisiert.<br />
2.9 CsCl-Gradientenzentrifugation<br />
Nach Transfektion viraler DNA in LMH-Zellen können in diesen nicht-infizierbaren Zellen die<br />
späten Schritte einer Infektion stattfinden. Das bedeutet, dass virale Proteine synthetisiert <strong>und</strong><br />
diese zu kompletten viralen Partikeln (Virionen <strong>und</strong> SVPs) zusammengesetzt werden.<br />
Zusätzlich zu diesen beiden Formen werden jedoch von den Zellen auch nicht-umhüllte Core-<br />
Partikel freigesetzt, die das virale Genom enthalten.<br />
Um Infektionsexperimente mit rekombinanten, aus den Kulturüberständen von<br />
transfizierten LMH-Zellen mit angereicherten Viren durchführen zu können, musste vorher die<br />
Menge der Virionen bestimmt werden. Dies geschah durch einen DNA-Dot Blot mit<br />
139
Material <strong>und</strong> Methoden<br />
vorhergehender CsCl-Gradientenzentrifugation. Die Gradientenzentrifugation war nötig, um die<br />
DNA-haltigen Virionen von den nicht-infektiösen Core-Partikeln abzutrennen.<br />
2.9.1 Anreicherung von Viren<br />
Vor der Gradientenzentrifugation wurden die viralen Partikel aus Zellkulturüberständen<br />
entweder mit PEG (Polyethylenglykol, Molekulargewicht 10.000, von Sigma, Deisenhofen)<br />
gefällt oder pelletiert, um sie zu konzentrieren. Dazu wurde zu dem Zellkulturüberstand 10 %<br />
PEG gegeben <strong>und</strong> das PEG sorgfältig aufgelöst. Dann wurden die Ansätze auf Eis für<br />
mindestens 2 h inkubiert, daraufhin wurde bei mindestens 4.000 rpm <strong>und</strong> 4°C für 1 h<br />
zentrifugiert. Das erhaltene Pellet enthielt nun die viralen Partikel <strong>und</strong> wurde in PBS oder<br />
Medium aufgenommen. Dabei wurde eine Konzentrierung von bis zu 100-fach erreicht.<br />
Alternativ wurden die viralen Partikel durch ein Kissen aus 500 µl 20 % Sukrose pelletiert. Dazu<br />
wurde der Kulturüberstand vorsichtig auf das Kissen aufgetragen <strong>und</strong> anschließend für 2 h bei<br />
38.000 rpm in der Ultrazentrifuge zentrifugiert. Der Überstand wurde abgenommen <strong>und</strong> das<br />
Pellet in PBS gelöst.<br />
2.9.2 CsCl-Gradientenzentrifugation<br />
Ein Aliquot der angereicherten Viren wurde in 2 ml Medium aufgenommen <strong>und</strong> auf einen<br />
Cäsiumchlorid-Gradienten aufgetragen. Dieser Gradient war wie folgt aufgebaut: am Gr<strong>und</strong>e<br />
des Zentrifugenröhrchens 0,5 ml 1,4 CsCl, darüber 0,5 ml 1,3 CsCl, darüber 0,5 ml 1,2 CsCl<br />
<strong>und</strong> darüber 0,5 ml 20 % Sukrose. Darauf wurde dann das virushaltige Medium aufgetragen.<br />
Die Auftrennung der verschiedenen viralen Partikel erfolgte durch diesen Stufengradienten in<br />
einem SW60-Rotor bei 20°C <strong>und</strong> 58.000 rpm für mindestens 3,5 h. Anschließend wurden<br />
verschiedene Fraktionen von unten nach oben mit einem ‚Fraction Recovery System’ von<br />
Beckman gesammelt. Insgesamt waren dies 12 Fraktionen. Diese Fraktionen wurden dann für<br />
den DNA -Dot Blot verwendet.<br />
2.9.3 DNA-Dot Blot<br />
Um die virale DNA in den einzelnen Fraktionen nachzuweisen, wurde die Methode des DNA-<br />
Dot Blot genutzt. Jede Fraktion wurde mit Hilfe einer Dot Blot-Apparatur auf eine Nylon-<br />
Membran (Hybond N von Amersham, Freiburg) aufgebracht. Außerdem wurden Standard-<br />
verdünnungen des DHBV-Plasmids DHBV 16t aufgetragen, um nach dem DNA-Nachweis die<br />
DNA-Menge in den Fraktionen genau berechnen zu können.<br />
Nach dem Dotten wurde die Membran getrocknet. Anschließend wurde die DNA auf der<br />
Membran denaturiert, indem diese zweimal 1,5 min auf einem mit Soak I getränkten<br />
Whatmanpapier inkubiert wurde. Dazwischen wurde sie gut getrocknet. Dann wurde<br />
140
Material <strong>und</strong> Methoden<br />
neutralisiert, indem die Membran viermal 1 min auf ein mit Soak II getränktes Whatmanpapier<br />
gelegt wurde. Diese Schritte wurden besonders vorsichtig durchgeführt, da die DNA auf der<br />
Membran noch nicht fixiert war. Nach einem weiteren Trocknungsschritt wurde die DNA auf der<br />
Membran durch eine Bestrahlung mit UV-Licht im UV -Stratalinker fixiert. Die Prähybridisierung<br />
erfolgte anschließend mit 5 ml QuickHyb (Stratagene, USA) für 30 min bei 68°C, dann wurde<br />
die denaturierte, radioaktive DNA-Sonde (siehe IV.2.10.2) zugegeben <strong>und</strong> über Nacht inkubiert.<br />
Am nächsten Tag wurde die Membran für 30 min mit Wash 1 bei 68°C gewaschen, dann für 30<br />
min bei RT mit Wash 2. Anschließend wurde sie exponiert <strong>und</strong> die Signale mit dem<br />
PhosphoImager quantifiziert. Die Auswertung erfolgte mit dem Programm TINA 2.0.<br />
Soak I: 0,5 M NaOH Soak II: 0,5 M Tris-HCl, pH 7,4<br />
1 M NaCl 3 M NaCl<br />
Wash 1: 10 % 20 x SSC Wash 2: 1 % 20 x SSC<br />
0,1 % SDS 0,5 % SDS<br />
2.10 Isolierung replikativer Intermediate <strong>und</strong> Southern Blot<br />
2.10.1 Isolierung replikativer Intermediate<br />
Um replikative Intermediate aus mit DHBV-Konstrukten transfizierten LMH-Zellen zu isolieren,<br />
wurden die Kulturen zuerst mit PBS gewaschen <strong>und</strong> dann von der Platte abgeschabt. Die<br />
Zellen wurden durch eine Zentrifugation für 5 min bei 4°C <strong>und</strong> 8.000 rpm pelletiert. Das Pellet<br />
wurde in 1 ml eines NP-40-haltigem Lysepuffer aufgenommen <strong>und</strong> lysiert. Dann wurde für 5 min<br />
bei 13.000 rpm zentrifugiert, um die Nuklei zu pelletieren. Der Überstand wurde in zwei<br />
Reaktionsgefäße überführt (je 500 µl), ein Gefäß wurde bei -20°C gelagert. Zu dem anderen<br />
wurden 100 µg/ml DNase I (Roche, Mannheim) gegeben, um die Input-DNA der Transfektion zu<br />
zerstören, virale DNA in Core-Partikeln war vor diesem Verdau geschützt. Es wurde für 30 min<br />
bei 37°C inkubiert, woraufhin 50 µl 0,5 M EDTA zugegeben <strong>und</strong> für weitere 5 min bei 37°C<br />
inkubiert wurde, um die DNase zu inaktivieren. Dann wurde die virale DNA freigesetzt, indem<br />
100 µg Proteinase K (Roche, Mannheim) zugegeben <strong>und</strong> 2 h bei 56°C inkubiert wurde.<br />
Anschließend wurde eine Phenol-Chloroform-Extraktion der DNA durchgeführt <strong>und</strong> danach die<br />
DNA mit Ethanol präzipitiert. Das daraus resultierende DNA -Pellet wurde in 20 µl H2O<br />
aufgenommen. 5 µl davon wurden für einen Southern Blot eingesetzt.<br />
NP-40-Puffer: 50 mM Tris-HCl, pH 8<br />
1 mM EDTA<br />
1 % Nonidet P-40<br />
141
2.10.2 Herstellung radioaktiv markierter Sonden<br />
Material <strong>und</strong> Methoden<br />
Eine DHBV-DNA-spezifische, radioaktive Sonde wurde für den Nachweis viraler DNA nach<br />
einem DNA-Dot Blot oder Southern Blot benötigt. Als Template für die Sondenherstellung<br />
diente das Plasmid DHBV 16t, das eine Tandem-Sequenz des vollständigen DHBV-Genoms<br />
enthielt. Von diesem Plasmid wurden 50 ng in TE -Puffer aufgenommen (Endvolumen 45 µl) <strong>und</strong><br />
diese erst für 5 min bei 99°C denaturiert <strong>und</strong> dann für 5 min auf Eis inkubiert. Die denaturierte<br />
DNA wurde in ein Reaktionsgefäß mit dem ‚Readyprime II Random Prime Labeling System’ von<br />
Amersham Pharmacia, Heidelberg, überführt. Nach Zugabe von 5 µl des radioaktiv markierten<br />
Nukleotids ( 32 P-dCTP) wurde der gesamte Ansatz gut gemischt <strong>und</strong> für 30 min bei 37°C<br />
inkubiert. Dabei wurde das markierte Nukleotid in kurze DNA -Stücke eingebaut, die<br />
komplementär zur DHBV-DNA waren. Anschließend wurde die Reaktion durch Zugabe von 5 µl<br />
0,2 M EDTA gestoppt. Um nicht eingebaute Nukleotide aus dem Ansatz zu entfernen, wurde er<br />
auf eine G25-Spin-Säule (Amersham, Heidelberg) gegeben <strong>und</strong> für 2 min bei 3.000 rpm<br />
zentrifugiert. Die Aktivität der Sonde wurde in einem Scintillation-Zähler bestimmt. Wenn diese<br />
ausreichend war, wurden 100 µl Heringssperma-DNA (Sigma, Deisenhofen) zugegeben, um<br />
unspezifische Bindungen der Sonde an die Membran zu verringern. Bevor die Sonde auf die<br />
Membran gegeben wurde, wurde sie bei 99°C für 5 min denaturiert.<br />
TE-Puffer: 10 mM Tris-HCl, pH 8,1<br />
2.10.3 Southern Blot<br />
1 mM EDTA<br />
Um die vorher isolierten replikativen Intermediate nachweisen zu können, wurde ein Southern<br />
Blot durchgeführt [199]. 5 µl der isolierten Intermediate wurden auf ein 1% Agarosegel<br />
aufgetragen <strong>und</strong> das Gel bei 40 V in 1 x TAE-Puffer über Nacht laufen gelassen. Nach dem<br />
Lauf wurde das Gel kurz mit ddH2O gewaschen <strong>und</strong> anschließend in 0,2 M Salzsäure für 30 min<br />
inkubiert, um die DNA zu depurinieren. Es folgte ein kurzer Waschschritt mit ddH2O, woraufhin<br />
die DNA durch dreißigminütige Inkubation in Denaturierungslösung denaturiert wurde. Dann<br />
wurde wieder kurz gewaschen <strong>und</strong> das Gel in Neutralisierungslösung 30 min inkubiert.<br />
Anschließend wurde das Gel kurz gewaschen <strong>und</strong> der Kapillarblot vorbereitet.<br />
Denaturierungslösung: 1,5 M NaCl<br />
0,5 M NaOH<br />
Neutralisierungslösung: 0,5 M Tris-HCl, pH 7,4<br />
3 M NaCl<br />
142
Material <strong>und</strong> Methoden<br />
Die Konstruktion für den Kapillarblot wurde wie folgt aufgebaut: In einer Schale befand<br />
sich 20 x SSC, darin wurde ein langes Stück Whatmanpapier getränkt <strong>und</strong> auf eine erhöhte<br />
Unterlage so gelegt, dass die Enden sich im Puffer befanden. Darauf wurden zwei<br />
angefeuchtete Stücke 3 MM Whatmanpapier gelegt, worauf das Gel folgte. Nachdem die<br />
Unterlage mit Parafilm abgedeckt war, wurde eine Nylonmembran (Amersham, Heidelberg) auf<br />
das Gel gelegt. Darauf folgten zwei trockene Stücke 3 MM Whatmanpapier <strong>und</strong> ein Stapel<br />
Papiertücher. Darauf wurde ein Gewicht gestellt (ca. 3 kg) <strong>und</strong> über Nacht geblottet. Der<br />
Transfer der DNA auf die Nylonmembran erfolgte dabei durch die Kapillarkräfte.<br />
Nach dem Kapillartransfer wurde die Membran kurz in 2 x SSC gewaschen <strong>und</strong> dann<br />
getrocknet. Anschließend wurde die DNA durch UV-Bestrahlung wie in Abschnitt IV.2.9.3<br />
beschrieben auf der Membran fixiert. Alle weiteren Schritte erfolgten wie in Abschnitt IV.2.9.3<br />
für den DNA-Dot Blot beschrieben.<br />
2.11 Vermehrung <strong>und</strong> Isolierung von Plasmid-DNA<br />
2.11.1 Transformation von Bakterien<br />
DNA wurde in kompetente Bakterien eingebracht, um eventuelle Strangbrüche zu reparieren<br />
<strong>und</strong> das Plasmid zu vermehren. Dazu wurden 50 µl Bakteriensuspension auf Eis aufgetaut <strong>und</strong><br />
mit 1 µg Plasmid-DNA vermischt. Der Ansatz wurde für 15 min auf Eis inkubiert, dann erfolgte<br />
der Hitzeschock für 90 sec bei 42°C. In dieser Zeit nahmen die Bakterien das an ihrer<br />
Oberfläche befindliche Plasmid auf. Es folgte eine Inkubation auf Eis für 2 min, um die Poren<br />
der bakteriellen Zellwand wieder zu schließen. Dann wurden 900 µl Standard I-Medium<br />
zugegeben <strong>und</strong> 30 min bei 37°C inkubiert. Die Bakteriensuspension wurde dann für 10 sec bei<br />
12.000 rpm zentrifugiert, um die Bakterien zu pelletieren. Das Pellet wurde in 30 µl Standard I-<br />
Medium aufgenommen <strong>und</strong> der gesamte Ansatz auf eine Selektionsplatte ausplattiert. Diese<br />
Agarplatte wurde über Nacht bei 37°C inkubiert. Resistente Klone, die das gewünschte Plasmid<br />
enthielten wurden für eine Plasmidpräparation in größerem Maßstab in 1 ml Flüssigmedium<br />
angeimpft.<br />
2.11.2 Maxipräparation von Plasmid-DNA<br />
Um große Mengen Plasmid-DNA für Transfektionen zu erhalten, wurden 250 ml Standard I-<br />
Medium mit dem entsprechenden Antibiotikum mit 1 ml Übernachtkultur angeimpft. Die<br />
Suspension wurde über Nacht bei 37°C <strong>und</strong> unter Schütteln inkubiert. Anschließend wurden die<br />
Bakterien bei 4°C <strong>und</strong> 6.000 rpm für 15 min durch Zentrifugation pelletiert. Das Pellet wurde<br />
dann in den entsprechenden Puffern aufgenommen <strong>und</strong> die DNA isoliert.<br />
143
Material <strong>und</strong> Methoden<br />
Die DNA wurde mit Hilfe des ‚Maxi Plasmid Isolation Kits’ von Qiagen isoliert <strong>und</strong> über<br />
eine Ionenaustauschsäule (Qiagen-Tip 500) nach den Angaben des Herstellers gereinigt.<br />
Anschließend wurde die DNA mit Isopropanol gefällt. Das Pellet wurde in 500 µl ddH2O<br />
aufgenommen <strong>und</strong> die Konzentration im Photometer bestimmt.<br />
144
V. Literatur<br />
Literatur<br />
1. Robinson, W.S., Hepatitis B Virus and Hepatitis Delta Virus. Principles and<br />
Practice of Infectious Diseases, ed. R. Gordon Douglas, G. Mandell, J. Bennett.<br />
1990, New York: Churchill Livingstone. 1204-1230.<br />
2. Blumberg, B.S., et al., A serum antigen (Australia antigen) in Down's syndrome,<br />
leukemia, and hepatitis. Ann Intern Med, 1967. 66(5): p. 924-31.<br />
3. Prince, A.M., An antigen detected in the blood during the incubation period of<br />
serum hepatitis. Proc Natl Acad Sci U S A, 1968. 60(3): p. 814-21.<br />
4. Dane, D.S., C.H. Cameron, and M. Briggs, Virus-like particles in serum of<br />
patients with Australia-antigen-associated hepatitis. Lancet, 1970. 1(7649): p.<br />
695-8.<br />
5. WHO, Fact Sheet Hepatitis B. Fact Sheets, 2000. 204.<br />
6. Hollinger, F.B. and T.J. Liang, Hepatitis B Virus, in Fields Virology, D.M. Knipe<br />
and P.M. Howley, Editors. 2001, Lippincott, Williams & Wilkins: Philadelphia. p.<br />
2971-3036.<br />
7. Lenhoff, R.J., C.A. Luscombe, and J. Summers, Competition in vivo between a<br />
cytopathic variant and a wild-type duck hepatitis B virus. Virology, 1998. 251(1):<br />
p. 85-95.<br />
8. Chisari, F.V. and C. Ferrari, Hepatitis B virus immunopathogenesis. Annu Rev<br />
Immunol, 1995. 13: p. 29-60.<br />
9. Moriyama, T., et al., Immunobiology and pathogenesis of hepatocellular injury<br />
in hepatitis B virus transgenic mice. Science, 1990. 248(4953): p. 361-4.<br />
10. Hoofnagle, J.H. and A.M. di Bisceglie, The treatment of chronic viral hepatitis. N<br />
Engl J Med, 1997. 336(5): p. 347-56.<br />
11. Kumar, R. and B. Agrawal, Novel treatment options for hepatitis B virus<br />
infection. Curr Opin Investig Drugs, 2004. 5(2): p. 171-8.<br />
12. Gust, I.D., et al., Taxonomic classification of human hepatitis B virus.<br />
Intervirology, 1986. 25(1): p. 14-29.<br />
13. Summers, J., J.M. Smolec, and R. Snyder, A virus similar to human hepatitis B<br />
virus associated with hepatitis and hepatoma in woodchucks. Proc Natl Acad<br />
Sci U S A, 1978. 75(9): p. 4533-7.<br />
14. Mason, W.S., G. Seal, and J. Summers, Virus of Pekin ducks with structural<br />
and biological relatedness to human hepatitis B virus. J Virol, 1980. 36(3): p.<br />
829-36.<br />
15. Sprengel, R., E.F. Kaleta, and H. Will, Isolation and characterization of a<br />
hepatitis B virus endemic in herons. J Virol, 1988. 62(10): p. 3832-9.<br />
16. Ganem, D. and R.J. Schneider, Hepadnaviridae: The viruses and their<br />
replication, in Fields Virology, D.M. Knipe and P.M. Howley, Editors. 2001,<br />
Lippincott, Williams & Wilkins: Philadelphia. p. 2923-2969.<br />
17. Tuttleman, J.S., J.C. Pugh, and J.W. Summers, In vitro experimental infection<br />
of primary duck hepatocyte cultures with duck hepatitis B virus. J Virol, 1986.<br />
58(1): p. 17-25.<br />
18. Urban, M.K., A.P. O'Connell, and W.T. London, Sequence of events in natural<br />
infection of Pekin duck embryos with duck hepatitis B virus. J Virol, 1985. 55(1):<br />
p. 16-22.<br />
19. Tagawa, M., W.S. Robinson, and P.L. Marion, Duck hepatitis B virus replicates<br />
in the yolk sac of developing embryos. J Virol, 1987. 61(7): p. 2273-9.<br />
20. Zhang, Y.Y. and J. Summers, Rapid production of neutralizing antibody leads to<br />
transient hepadnavirus infection. J Virol, 2004. 78(3): p. 1195-201.<br />
21. Freiman, J.S., et al., Experimental duck hepatitis B virus infection: pathology<br />
and evolution of hepatic and extrahepatic infection. Hepatology, 1988. 8(3): p.<br />
507-13.<br />
145
Literatur<br />
22. Gavilanes, F., J.M. Gonzalez-Ros, and D.L. Peterson, Structure of hepatitis B<br />
surface antigen. Characterization of the lipid components and their association<br />
with the viral proteins. J Biol Chem, 1982. 257(13): p. 7770-7.<br />
23. Sonveaux, N., D. Thines, and J.M. Ruysschaert, Characterization of the HBsAg<br />
particle lipid membrane. Res Virol, 1995. 146(1): p. 43-51.<br />
24. Satoh, O., et al., Membrane structure of the hepatitis B virus surface antigen<br />
particle. J Biochem (Tokyo), 2000. 127(4): p. 543-50.<br />
25. Klingmuller, U. and H. Schaller, Hepadnavirus infection requires interaction<br />
between the viral pre-S domain and a specific hepatocellular receptor. J Virol,<br />
1993. 67(12): p. 7414-22.<br />
26. Bruns, M., et al., Enhancement of hepatitis B virus infection by noninfectious<br />
subviral particles. J Virol, 1998. 72(2): p. 1462-8.<br />
27. Nassal, M. and H. Schaller, Hepatitis B virus replication. Trends Microbiol,<br />
1993. 1(6): p. 221-8.<br />
28. Hildt, M., Zelluläre Funktionen während der frühen <strong>und</strong> späten Schritte im<br />
Infektionszyklus des Enten Hepatitis B Virus. Dissertation der<br />
Naturwissenschaftlich-Mathematischen Gesamtfakultät der Ruprecht-Karls-<br />
Universität Heidelberg, 1996.<br />
29. Lien, J.M., et al., Initiation and termination of duck hepatitis B virus DNA<br />
synthesis during virus maturation. J Virol, 1987. 61(12): p. 3832-40.<br />
30. Gerlich, W.H. and W.S. Robinson, Hepatitis B virus contains protein attached to<br />
the 5' terminus of its complete DNA strand. Cell, 1980. 21(3): p. 801-9.<br />
31. Bosch, V., et al., The duck hepatitis B virus P-gene codes for protein strongly<br />
associated with the 5'-end of the viral DNA minus strand. Virology, 1988.<br />
166(2): p. 475-85.<br />
32. Landers, T.A., H.B. Greenberg, and W.S. Robinson, Structure of hepatitis B<br />
Dane particle DNA and nature of the endogenous DNA polymerase reaction. J<br />
Virol, 1977. 23(2): p. 368-76.<br />
33. Seeger, C., D. Ganem, and H.E. Varmus, Biochemical and genetic evidence for<br />
the hepatitis B virus replication strategy. Science, 1986. 232(4749): p. 477-84.<br />
34. Seeger, C., D. Ganem, and H.E. Varmus, Nucleotide sequence of an infectious<br />
molecularly cloned genome of gro<strong>und</strong> squirrel hepatitis virus. J Virol, 1984.<br />
51(2): p. 367-75.<br />
35. Chang, S.F., et al., Duck hepatitis B virus expresses a regulatory HBx-like<br />
protein from a hidden open reading frame. J Virol, 2001. 75(1): p. 161-70.<br />
36. Su, Q., et al., Circulating hepatitis B virus nucleic acids in chronic infection :<br />
representation of differently polyadenylated viral transcripts during progression<br />
to nonreplicative stages. Clin Cancer Res, 2001. 7(7): p. 2005-15.<br />
37. Yaginuma, K., et al., Hepatitis B virus (HBV) particles are produced in a cell<br />
culture system by transient expression of transfected HBV DNA. Proc Natl Acad<br />
Sci U S A, 1987. 84(9): p. 2678-82.<br />
38. Kock, J. and H.J. Schlicht, Analysis of the earliest steps of hepadnavirus<br />
replication: genome repair after infectious entry into hepatocytes does not<br />
depend on viral polymerase activity. J Virol, 1993. 67(8): p. 4867-74.<br />
39. Bock, C.T., et al., Hepatitis B virus genome is organized into nucleosomes in<br />
the nucleus of the infected cell. Virus Genes, 1994. 8(3): p. 215-29.<br />
40. Newbold, J.E., et al., The covalently closed duplex form of the hepadnavirus<br />
genome exists in situ as a heterogeneous population of viral minichromosomes.<br />
J Virol, 1995. 69(6): p. 3350-7.<br />
41. Feitelson, M.A., P.L. Marion, and W.S. Robinson, Core particles of hepatitis B<br />
virus and gro<strong>und</strong> squirrel hepatitis virus. II. Characterization of the protein<br />
kinase reaction associated with gro<strong>und</strong> squirrel hepatitis virus and hepatitis B<br />
virus. J Virol, 1982. 43(2): p. 741-8.<br />
42. Beames, B. and R.E. Lanford, Carboxy-terminal truncations of the HBV core<br />
protein affect capsid formation and the apparent size of encapsidated HBV<br />
RNA. Virology, 1993. 194(2): p. 597-607.<br />
146
Literatur<br />
43. Mabit, H., et al., Signals for bidirectional nucleocytoplasmic transport in the<br />
duck hepatitis B virus capsid protein. J Virol, 2001. 75(4): p. 1968-77.<br />
44. Pollack, J.R. and D. Ganem, An RNA stem-loop structure directs hepatitis B<br />
virus genomic RNA encapsidation. J Virol, 1993. 67(6): p. 3254-63.<br />
45. Garcia, P.D., et al., Targeting of the hepatitis B virus precore protein to the<br />
endoplasmic reticulum membrane: after signal peptide cleavage translocation<br />
can be aborted and the product released into the cytoplasm. J Cell Biol, 1988.<br />
106(4): p. 1093-104.<br />
46. Schlicht, H.J., J. Salfeld, and H. Schaller, The duck hepatitis B virus pre-C<br />
region encodes a signal sequence which is essential for synthesis and<br />
secretion of processed core proteins but not for virus formation. J Virol, 1987.<br />
61(12): p. 3701-9.<br />
47. Schneider, R., et al., Mechanism, kinetics, and role of duck hepatitis B virus eantigen<br />
expression in vivo. Virology, 1991. 182(2): p. 503-12.<br />
48. Pugh, J.C., et al., Characterization of a pre-S polypeptide on the surfaces of<br />
infectious avian hepadnavirus particles. J Virol, 1987. 61(5): p. 1384-90.<br />
49. Bruss, V., et al., Functions of the large hepatitis B virus surface protein in viral<br />
particle morphogenesis. Intervirology, 1996. 39(1-2): p. 23-31.<br />
50. Schlicht, H.J., et al., Biochemical and immunological characterization of the<br />
duck hepatitis B virus envelope proteins. J Virol, 1987. 61(7): p. 2280-5.<br />
51. Sprengel, R., et al., Comparative sequence analysis of duck and human<br />
hepatitis B virus genomes. J Med Virol, 1985. 15(4): p. 323-33.<br />
52. Macrae, D.R., V. Bruss, and D. Ganem, Myristylation of a duck hepatitis B virus<br />
envelope protein is essential for infectivity but not for virus assembly. Virology,<br />
1991. 181(1): p. 359-63.<br />
53. Bruss, V., et al., Post-translational alterations in transmembrane topology of the<br />
hepatitis B virus large envelope protein. Embo J, 1994. 13(10): p. 2273-9.<br />
54. Lambert, C. and R. Prange, Dual topology of the hepatitis B virus large<br />
envelope protein: determinants influencing post-translational pre-S<br />
translocation. J Biol Chem, 2001. 276(25): p. 22265-72. Epub 2001 Apr 11.<br />
55. Kaplan, P.M., et al., DNA polymerase associated with human hepatitis B<br />
antigen. J Virol, 1973. 12(5): p. 995-1005.<br />
56. Radziwill, G., W. Tucker, and H. Schaller, Mutational analysis of the hepatitis B<br />
virus P gene product: domain structure and RNase H activity. J Virol, 1990.<br />
64(2): p. 613-20.<br />
57. Summers, J. and W.S. Mason, Replication of the genome of a hepatitis B--like<br />
virus by reverse transcription of an RNA intermediate. Cell, 1982. 29(2): p. 403-<br />
15.<br />
58. Meier, P., et al., A duck hepatitis B virus strain with a knockout mutation in the<br />
putative X ORF shows similar infectivity and in vivo growth characteristics to<br />
wild-type virus. Virology, 2003. 317(2): p. 291-8.<br />
59. Twu, J.S. and R.H. Schloemer, Transcriptional trans-activating function of<br />
hepatitis B virus. J Virol, 1987. 61(11): p. 3448-53.<br />
60. Kock, J., E.M. Borst, and H.J. Schlicht, Uptake of duck hepatitis B virus into<br />
hepatocytes occurs by endocytosis but does not require passage of the virus<br />
through an acidic intracellular compartment. J Virol, 1996. 70(9): p. 5827-31.<br />
61. Rodriguez-Crespo, I., et al., Fusogenic activity of hepadnavirus peptides<br />
corresponding to sequences downstream of the putative cleavage site.<br />
Virology, 1999. 261(1): p. 133-42.<br />
62. Kann, M., et al., Phosphorylation-dependent binding of hepatitis B virus core<br />
particles to the nuclear pore complex. J Cell Biol, 1999. 145(1): p. 45-55.<br />
63. Wang, J., et al., Hepatitis B virus integration in a cyclin A gene in a<br />
hepatocellular carcinoma. Nature, 1990. 343(6258): p. 555-7.<br />
64. Rall, L.B., et al., Transcription of hepatitis B virus by RNA polymerase II. Mol<br />
Cell Biol, 1983. 3(10): p. 1766-73.<br />
147
Literatur<br />
65. Eble, B.E., V.R. Lingappa, and D. Ganem, The N-terminal (pre-S2) domain of a<br />
hepatitis B virus surface glycoprotein is translocated across membranes by<br />
downstream signal sequences. J Virol, 1990. 64(3): p. 1414-9.<br />
66. Zhou, S. and D.N. Standring, Hepatitis B virus capsid particles are assembled<br />
from core-protein dimer precursors. Proc Natl Acad Sci U S A, 1992. 89(21): p.<br />
10046-50.<br />
67. Gazina, E.V., et al., Core protein phosphorylation modulates pregenomic RNA<br />
encapsidation to different extents in human and duck hepatitis B viruses. J<br />
Virol, 2000. 74(10): p. 4721-8.<br />
68. Pugh, J., A. Zweidler, and J. Summers, Characterization of the major duck<br />
hepatitis B virus core particle protein. J Virol, 1989. 63(3): p. 1371-6.<br />
69. Zhang, Y.Y., et al., Single-cell analysis of covalently closed circular DNA copy<br />
numbers in a hepadnavirus-infected liver. Proc Natl Acad Sci U S A, 2003.<br />
100(21): p. 12372-7. Epub 2003 Oct 3.<br />
70. Lenhoff, R.J. and J. Summers, Coordinate regulation of replication and virus<br />
assembly by the large envelope protein of an avian hepadnavirus. J Virol, 1994.<br />
68(7): p. 4565-71.<br />
71. Petit, M.A., et al., PreS1-specific binding proteins as potential receptors for<br />
hepatitis B virus in human hepatocytes. Virology, 1992. 187(1): p. 211-22.<br />
72. Treichel, U., et al., The asialoglycoprotein receptor mediates hepatic binding<br />
and uptake of natural hepatitis B virus particles derived from viraemic carriers. J<br />
Gen Virol, 1994. 75(Pt 11): p. 3021-9.<br />
73. Urban, S. and P. Gripon, Inhibition of duck hepatitis B virus infection by a<br />
myristoylated pre-S peptide of the large viral surface protein. J Virol, 2002.<br />
76(4): p. 1986-90.<br />
74. Urban, S., et al., Avian hepatitis B virus infection is initiated by the interaction of<br />
a distinct pre-S subdomain with the cellular receptor gp180. J Virol, 1998.<br />
72(10): p. 8089-97.<br />
75. Tong, S., J. Li, and J.R. Wands, Carboxypeptidase D is an avian hepatitis B<br />
virus receptor. J Virol, 1999. 73(10): p. 8696-702.<br />
76. Urban, S., et al., Receptor recognition by a hepatitis B virus reveals a novel<br />
mode of high affinity virus-receptor interaction. Embo J, 2000. 19(6): p. 1217-<br />
27.<br />
77. Song, L. and L.D. Fricker, Tissue distribution and characterization of soluble<br />
and membrane-bo<strong>und</strong> forms of metallocarboxypeptidase D. J Biol Chem, 1996.<br />
271(46): p. 28884-9.<br />
78. Pugh, J.C. and J.W. Summers, Infection and uptake of duck hepatitis B virus by<br />
duck hepatocytes maintained in the presence of dimethyl sulfoxide. Virology,<br />
1989. 172(2): p. 564-72.<br />
79. Rigg, R.J. and H. Schaller, Duck hepatitis B virus infection of hepatocytes is not<br />
dependent on low pH. J Virol, 1992. 66(5): p. 2829-36.<br />
80. Offensperger, W.B., et al., Inhibition of duck hepatitis B virus infection by<br />
lysosomotropic agents. Virology, 1991. 183(1): p. 415-8.<br />
81. Breiner, K.M. and H. Schaller, Cellular receptor traffic is essential for productive<br />
duck hepatitis B virus infection. J Virol, 2000. 74(5): p. 2203-9.<br />
82. Lu, X., T.M. Block, and W.H. Gerlich, Protease-induced infectivity of hepatitis B<br />
virus for a human hepatoblastoma cell line. J Virol, 1996. 70(4): p. 2277-85.<br />
83. Goldstein, J.L., R.G. Anderson, and M.S. Brown, Coated pits, coated vesicles,<br />
and receptor-mediated endocytosis. Nature, 1979. 279(5715): p. 679-85.<br />
84. Maxfield, F.R. and T.E. McGraw, Endocytic recycling. Nat Rev Mol Cell Biol,<br />
2004. 5(2): p. 121-32.<br />
85. Araki, N., M.T. Johnson, and J.A. Swanson, A role for phosphoinositide 3kinase<br />
in the completion of macropinocytosis and phagocytosis by<br />
macrophages. J Cell Biol, 1996. 135(5): p. 1249-60.<br />
86. Parton, R.G., Caveolae--from ultrastructure to molecular mechanisms. Nat Rev<br />
Mol Cell Biol, 2003. 4(2): p. 162-7.<br />
148
Literatur<br />
87. Thomsen, P., et al., Caveolae are highly immobile plasma membrane<br />
microdomains, which are not involved in constitutive endocytic trafficking. Mol<br />
Biol Cell, 2002. 13(1): p. 238-50.<br />
88. Morrissette, N., E. Gold, and A. Aderem, The macrophage--a cell for all<br />
seasons. Trends Cell Biol, 1999. 9(5): p. 199-201.<br />
89. Myers, J.N., et al., Beta-very low density lipoprotein is sequestered in surfaceconnected<br />
tubules in mouse peritoneal macrophages. J Cell Biol, 1993. 123(6<br />
Pt 1): p. 1389-402.<br />
90. Sandvig, K. and B. van Deurs, Membrane traffic exploited by protein toxins.<br />
Annu Rev Cell Dev Biol, 2002. 18: p. 1-24. Epub 2002 Apr 2.<br />
91. Pakdaman, R. and J.M. El Hage Chahine, A mechanism for iron uptake by<br />
transferrin. Eur J Biochem, 1996. 236(3): p. 922-31.<br />
92. Chang, M.P., et al., Adaptor self-aggregation, adaptor-receptor recognition and<br />
binding of alpha-adaptin subunits to the plasma membrane contribute to<br />
recruitment of adaptor (AP2) components of clathrin-coated pits. Embo J, 1993.<br />
12(5): p. 2169-80.<br />
93. Ungewickell, E. and D. Branton, Assembly units of clathrin coats. Nature, 1981.<br />
289(5796): p. 420-2.<br />
94. Cremona, O. and P. De Camilli, Synaptic vesicle endocytosis. Curr Opin<br />
Neurobiol, 1997. 7(3): p. 323-30.<br />
95. Takei, K., et al., Tubular membrane invaginations coated by dynamin rings are<br />
induced by GTP-gamma S in nerve terminals. Nature, 1995. 374(6518): p. 186-<br />
90.<br />
96. Jones, S.M., et al., Role of dynamin in the formation of transport vesicles from<br />
the trans-Golgi network. Science, 1998. 279(5350): p. 573-7.<br />
97. Wilson, J.M., et al., EEA1, a tethering protein of the early sorting endosome,<br />
shows a polarized distribution in hippocampal neurons, epithelial cells, and<br />
fibroblasts. Mol Biol Cell, 2000. 11(8): p. 2657-71.<br />
98. Gorvel, J.P., et al., rab5 controls early endosome fusion in vitro. Cell, 1991.<br />
64(5): p. 915-25.<br />
99. Gruenberg, J. and K.E. Howell, Membrane traffic in endocytosis: insights from<br />
cell-free assays. Annu Rev Cell Biol, 1989. 5: p. 453-81.<br />
100. Mellman, I., R. Fuchs, and A. Helenius, Acidification of the endocytic and<br />
exocytic pathways. Annu Rev Biochem, 1986. 55: p. 663-700.<br />
101. Aniento, F., et al., Cytoplasmic dynein-dependent vesicular transport from early<br />
to late endosomes. J Cell Biol, 1993. 123(6 Pt 1): p. 1373-87.<br />
<strong>102</strong>. Sorkin, A. and M. Von Zastrow, Signal transduction and endocytosis: close<br />
encounters of many kinds. Nat Rev Mol Cell Biol, 2002. 3(8): p. 600-14.<br />
103. Grabe, M. and G. Oster, Regulation of organelle acidity. J Gen Physiol, 2001.<br />
117(4): p. 329-44.<br />
104. Al-Awqati, Q., Proton-translocating ATPases. Annu Rev Cell Biol, 1986. 2: p.<br />
179-99.<br />
105. Gruenberg, J. and F.R. Maxfield, Membrane transport in the endocytic pathway.<br />
Curr Opin Cell Biol, 1995. 7(4): p. 552-63.<br />
106. Enrich, C., G. Apodaca, and K.E. Mostov, Calmodulin regulates the intracellular<br />
trafficking in epithelial cells. Z Gastroenterol, 1996. 34(Suppl 3): p. 83-5.<br />
107. Bretscher, M.S. and S. Munro, Cholesterol and the Golgi apparatus. Science,<br />
1993. 261(5126): p. 1280-1.<br />
108. Schroeder, F. and G. Nemecz, Transmembrane cholesterol distribution.<br />
Advances in Cholesterol Research, ed. M. Esfahani and J. Swaney. 1990,<br />
Caldwell, NJ: Telford Press. 47-87.<br />
109. Brown, D.A. and E. London, Functions of lipid rafts in biological membranes.<br />
Annu Rev Cell Dev Biol, 1998. 14: p. 111-36.<br />
110. Brown, D.A. and E. London, Structure and function of sphingolipid- and<br />
cholesterol-rich membrane rafts. J Biol Chem, 2000. 275(23): p. 17221-4.<br />
149
Literatur<br />
111. Schnitzer, J.E., et al., Filipin-sensitive caveolae-mediated transport in<br />
endothelium: reduced transcytosis, scavenger endocytosis, and capillary<br />
permeability of select macromolecules. J Cell Biol, 1994. 127(5): p. 1217-32.<br />
112. Anderson, R.G., The caveolae membrane system. Annu Rev Biochem, 1998.<br />
67: p. 199-225.<br />
113. Calvo, M., et al., Morphologic and functional characterization of caveolae in rat<br />
liver hepatocytes. Hepatology, 2001. 33(5): p. 1259-69.<br />
114. Kartenbeck, J., H. Stukenbrok, and A. Helenius, Endocytosis of simian virus 40<br />
into the endoplasmic reticulum. J Cell Biol, 1989. 109(6 Pt 1): p. 2721-9.<br />
115. Montesano, R., et al., Non-coated membrane invaginations are involved in<br />
binding and internalization of cholera and tetanus toxins. Nature, 1982.<br />
296(5858): p. 651-3.<br />
116. Thiele, C., et al., Cholesterol binds to synaptophysin and is required for<br />
biogenesis of synaptic vesicles. Nat Cell Biol, 2000. 2(1): p. 42-9.<br />
117. Murata, M., et al., VIP21/caveolin is a cholesterol-binding protein. Proc Natl<br />
Acad Sci U S A, 1995. 92(22): p. 10339-43.<br />
118. Monier, S., et al., Oligomerization of VIP21-caveolin in vitro is stabilized by long<br />
chain fatty acylation or cholesterol. FEBS Lett, 1996. 388(2-3): p. 143-9.<br />
119. Song, K.S., et al., Mutational analysis of the properties of caveolin-1. A novel<br />
role for the C-terminal domain in mediating homo-typic caveolin-caveolin<br />
interactions. J Biol Chem, 1997. 272(7): p. 4398-403.<br />
120. Rothberg, K.G., et al., Caveolin, a protein component of caveolae membrane<br />
coats. Cell, 1992. 68(4): p. 673-82.<br />
121. Majoul, I., et al., KDEL receptor (Erd2p)-mediated retrograde transport of the<br />
cholera toxin A subunit from the Golgi involves COPI, p23, and the COOH<br />
terminus of Erd2p. J Cell Biol, 1998. 143(3): p. 601-12.<br />
122. Rawat, S.S., et al., Modulation of entry of enveloped viruses by cholesterol and<br />
sphingolipids (Review). Mol Membr Biol, 2003. 20(3): p. 243-54.<br />
123. Stein, B.S., et al., pH-independent HIV entry into CD4-positive T cells via virus<br />
envelope fusion to the plasma membrane. Cell, 1987. 49(5): p. 659-68.<br />
124. White, J., K. Matlin, and A. Helenius, Cell fusion by Semliki Forest, influenza,<br />
and vesicular stomatitis viruses. J Cell Biol, 1981. 89(3): p. 674-9.<br />
125. Bayer, N., et al., Human rhinovirus HRV14 uncoats from early endosomes in<br />
the presence of bafilomycin. FEBS Lett, 1999. 463(1-2): p. 175-8.<br />
126. Bayer, N., et al., Effect of bafilomycin A1 and nocodazole on endocytic transport<br />
in HeLa cells: implications for viral uncoating and infection. J Virol, 1998.<br />
72(12): p. 9645-55.<br />
127. Coppens, I. and K.A. Joiner, Host but not parasite cholesterol controls<br />
Toxoplasma cell entry by modulating organelle discharge. Mol Biol Cell, 2003.<br />
14(9): p. 3804-20. Epub 2003 May 29.<br />
128. Manes, S., et al., Membrane raft microdomains mediate lateral assemblies<br />
required for HIV-1 infection. EMBO Rep, 2000. 1(2): p. 190-6.<br />
129. Anderson, H.A., Y. Chen, and L.C. Norkin, Bo<strong>und</strong> simian virus 40 translocates<br />
to caveolin-enriched membrane domains, and its entry is inhibited by drugs that<br />
selectively disrupt caveolae. Mol Biol Cell, 1996. 7(11): p. 1825-34.<br />
130. Pelkmans, L., J. Kartenbeck, and A. Helenius, Caveolar endocytosis of simian<br />
virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell<br />
Biol, 2001. 3(5): p. 473-83.<br />
131. Giranda, V.L., et al., Acid-induced structural changes in human rhinovirus 14:<br />
possible role in uncoating. Proc Natl Acad Sci U S A, 1992. 89(21): p. <strong>102</strong>13-7.<br />
132. Kim, S., et al., Conformational variability of a picornavirus capsid: pHdependent<br />
structural changes of Mengo virus related to its host receptor<br />
attachment site and disassembly. Virology, 1990. 175(1): p. 176-90.<br />
133. Skehel, J.J. and D.C. Wiley, Receptor binding and membrane fusion in virus<br />
entry: the influenza hemagglutinin. Annu Rev Biochem, 2000. 69: p. 531-69.<br />
150
Literatur<br />
134. Lescar, J., et al., The Fusion glycoprotein shell of Semliki Forest virus: an<br />
icosahedral assembly primed for fusogenic activation at endosomal pH. Cell,<br />
2001. 105(1): p. 137-48.<br />
135. Colman, P.M. and M.C. Lawrence, The structural biology of type I viral<br />
membrane fusion. Nat Rev Mol Cell Biol, 2003. 4(4): p. 309-19.<br />
136. Endow, S.A., Kinesin motors as molecular machines. Bioessays, 2003. 25(12):<br />
p. 1212-9.<br />
137. Vallee, R.B., et al., Dynein: An ancient motor protein involved in multiple modes<br />
of transport. J Neurobiol, 2004. 58(2): p. 189-200.<br />
138. Gottlieb, T.A., et al., Actin microfilaments play a critical role in endocytosis at<br />
the apical but not the basolateral surface of polarized epithelial cells. J Cell Biol,<br />
1993. 120(3): p. 695-710.<br />
139. Martin, K. and A. Helenius, Transport of incoming influenza virus nucleocapsids<br />
into the nucleus. J Virol, 1991. 65(1): p. 232-44.<br />
140. Iyengar, S., J.E. Hildreth, and D.H. Schwartz, Actin-dependent receptor<br />
colocalization required for human immunodeficiency virus entry into host cells. J<br />
Virol, 1998. 72(6): p. 5251-5.<br />
141. Smith, G.A. and L.W. Enquist, Break ins and break outs: viral interactions with<br />
the cytoskeleton of Mammalian cells. Annu Rev Cell Dev Biol, 2002. 18: p. 135-<br />
61. Epub 2002 Apr 2.<br />
142. Sodeik, B., M.W. Ebersold, and A. Helenius, Microtubule-mediated transport of<br />
incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol, 1997.<br />
136(5): p. 1007-21.<br />
143. Topp, K.S., L.B. Meade, and J.H. LaVail, Microtubule polarity in the peripheral<br />
processes of trigeminal ganglion cells: relevance for the retrograde transport of<br />
herpes simplex virus. J Neurosci, 1994. 14(1): p. 318-25.<br />
144. Sodeik, B., Unchain my heart, baby let me go--the entry and intracellular<br />
transport of HIV. J Cell Biol, 2002. 159(3): p. 393-5. Epub 2002 Nov 11.<br />
145. Schwartz, O., et al., Antiviral activity of the proteasome on incoming human<br />
immunodeficiency virus type 1. J Virol, 1998. 72(5): p. 3845-50.<br />
146. Damsky, C.H., et al., Is there a role for actin in virus budding? J Cell Biol, 1977.<br />
75(2 Pt 1): p. 593-605.<br />
147. Miranda-Saksena, M., et al., Anterograde transport of herpes simplex virus type<br />
1 in cultured, dissociated human and rat dorsal root ganglion neurons. J Virol,<br />
2000. 74(4): p. 1827-39.<br />
148. Dingwell, K.S., et al., Herpes simplex virus glycoproteins E and I facilitate cellto-cell<br />
spread in vivo and across junctions of cultured cells. J Virol, 1994. 68(2):<br />
p. 834-45.<br />
149. Phillips, D.M. and A.S. Bourinbaiar, Mechanism of HIV spread from<br />
lymphocytes to epithelia. Virology, 1992. 186(1): p. 261-73.<br />
150. Wolffe, E.J., et al., The A34R glycoprotein gene is required for induction of<br />
specialized actin-containing microvilli and efficient cell-to-cell transmission of<br />
vaccinia virus. J Virol, 1997. 71(5): p. 3904-15.<br />
151. Igakura, T., et al., Spread of HTLV-I between lymphocytes by virus-induced<br />
polarization of the cytoskeleton. Science, 2003. 299(5613): p. 1713-6. Epub<br />
2003 Feb 13.<br />
152. Law, M., R. Hollinshead, and G.L. Smith, Antibody-sensitive and antibodyresistant<br />
cell-to-cell spread by vaccinia virus: role of the A33R protein in<br />
antibody-resistant spread. J Gen Virol, 2002. 83(Pt 1): p. 209-22.<br />
153. Offensperger, W.B., et al., Suramin prevents duck hepatitis B virus infection in<br />
vivo. Antimicrob Agents Chemother, 1993. 37(7): p. 1539-42.<br />
154. Petcu, D.J., et al., Suramin inhibits in vitro infection by duck hepatitis B virus,<br />
Rous sarcoma virus, and hepatitis delta virus. Virology, 1988. 167(2): p. 385-92.<br />
155. Aguilar, J.S., M. Rice, and E.K. Wagner, The polysulfonated compo<strong>und</strong> suramin<br />
blocks adsorption and lateral difusion of herpes simplex virus type-1 in vero<br />
cells. Virology, 1999. 258(1): p. 141-51.<br />
151
Literatur<br />
156. Yahi, N., et al., Suramin inhibits binding of the V3 region of HIV-1 envelope<br />
glycoprotein gp120 to galactosylceramide, the receptor for HIV-1 gp120 on<br />
human colon epithelial cells. J Biol Chem, 1994. 269(39): p. 24349-53.<br />
157. Irurzun, A., J.L. Nieva, and L. Carrasco, Entry of Semliki forest virus into cells:<br />
effects of concanamycin A and nigericin on viral membrane fusion and infection.<br />
Virology, 1997. 227(2): p. 488-92.<br />
158. Svensson, U. and R. Persson, Entry of adenovirus 2 into HeLa cells. J Virol,<br />
1984. 51(3): p. 687-94.<br />
159. Ploubidou, A. and M. Way, Viral transport and the cytoskeleton. Curr Opin Cell<br />
Biol, 2001. 13(1): p. 97-105.<br />
160. Sodeik, B., Mechanisms of viral transport in the cytoplasm. Trends Microbiol,<br />
2000. 8(10): p. 465-72.<br />
161. Summers, J., P.M. Smith, and A.L. Horwich, Hepadnavirus envelope proteins<br />
regulate covalently closed circular DNA amplification. J Virol, 1990. 64(6): p.<br />
2819-24.<br />
162. Funk, A., Studien zu Aufnahme <strong>und</strong> intrazellulärem Transport von Hepatitis B-<br />
Viren. Diplomarbeit, 2002. TU Darmstadt.<br />
163. Mabit, H., et al., Intact microtubules support adenovirus and herpes simplex<br />
virus infections. J Virol, 2002. 76(19): p. 9962-71.<br />
164. Gilbert, J.M., I.G. Goldberg, and T.L. Benjamin, Cell penetration and trafficking<br />
of polyomavirus. J Virol, 2003. 77(4): p. 2615-22.<br />
165. Liu, W.J., et al., Association of bovine papillomavirus type 1 with microtubules.<br />
Virology, 2001. 282(2): p. 237-44.<br />
166. Khawaja, S., G.G. G<strong>und</strong>ersen, and J.C. Bulinski, Enhanced stability of<br />
microtubules enriched in detyrosinated tubulin is not a direct function of<br />
detyrosination level. J Cell Biol, 1988. 106(1): p. 141-9.<br />
167. Ogawa-Goto, K., et al., Microtubule network facilitates nuclear targeting of<br />
human cytomegalovirus capsid. J Virol, 2003. 77(15): p. 8541-7.<br />
168. Bukrinskaya, A., et al., Establishment of a functional human immunodeficiency<br />
virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J<br />
Exp Med, 1998. 188(11): p. 2113-25.<br />
169. Satoh, O., et al., Lipid composition of hepatitis B virus surface antigen particles<br />
and the particle-producing human hepatoma cell lines. J Lipid Res, 1990. 31(7):<br />
p. 1293-300.<br />
170. Empig, C.J. and M.A. Goldsmith, Association of the caveola vesicular system<br />
with cellular entry by filoviruses. J Virol, 2002. 76(10): p. 5266-70.<br />
171. Werling, D., et al., Involvement of caveolae in the uptake of respiratory syncytial<br />
virus antigen by dendritic cells. J Leukoc Biol, 1999. 66(1): p. 50-8.<br />
172. Kilsdonk, E.P., et al., Cellular cholesterol efflux mediated by cyclodextrins. J<br />
Biol Chem, 1995. 270(29): p. 17250-6.<br />
173. Gent, M.P. and J.H. Prestegard, Interaction of the polyene antibiotics with lipid<br />
bilayer vesicles containing cholesterol. Biochim Biophys Acta, 1976. 426(1): p.<br />
17-30.<br />
174. Lencer, W.I., T.R. Hirst, and R.K. Holmes, Membrane traffic and the cellular<br />
uptake of cholera toxin. Biochim Biophys Acta, 1999. 1450(3): p. 177-90.<br />
175. Alberts, A.W., et al., Mevinolin: a highly potent competitive inhibitor of<br />
hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent.<br />
Proc Natl Acad Sci U S A, 1980. 77(7): p. 3957-61.<br />
176. Foster, L.J., C.L. De Hoog, and M. Mann, Unbiased quantitative proteomics of<br />
lipid rafts reveals high specificity for signaling factors. Proc Natl Acad Sci U S A,<br />
2003. 100(10): p. 5813-8. Epub 2003 Apr 30.<br />
177. Shogomori, H. and A.H. Futerman, Cholesterol depletion by methyl-betacyclodextrin<br />
blocks cholera toxin transport from endosomes to the Golgi<br />
apparatus in hippocampal neurons. J Neurochem, 2001. 78(5): p. 991-9.<br />
152
Literatur<br />
178. Rodal, S.K., et al., Extraction of cholesterol with methyl-beta-cyclodextrin<br />
perturbs formation of clathrin-coated endocytic vesicles. Mol Biol Cell, 1999.<br />
10(4): p. 961-74.<br />
179. Guyader, M., et al., Role for human immunodeficiency virus type 1 membrane<br />
cholesterol in viral internalization. J Virol, 2002. 76(20): p. 10356-64.<br />
180. Campbell, S.M., S.M. Crowe, and J. Mak, Virion-associated cholesterol is<br />
critical for the maintenance of HIV-1 structure and infectivity. Aids, 2002.<br />
16(17): p. 2253-61.<br />
181. Oess, S. and E. Hildt, Novel cell permeable motif derived from the PreS2domain<br />
of hepatitis-B virus surface antigens. Gene Ther, 2000. 7(9): p. 750-8.<br />
182. Saher, G. and E. Hildt, Activation of c-Raf-1 kinase signal transduction pathway<br />
in alpha(7) integrin-deficient mice. J Biol Chem, 1999. 274(39): p. 27651-7.<br />
183. Kyte, J. and R.F. Doolittle, A simple method for displaying the hydropathic<br />
character of a protein. J Mol Biol, 1982. 157(1): p. 105-32.<br />
184. Fernholz, D., Studien zur Synthese <strong>und</strong> Funktion der Hüllproteine bei Hepatitis<br />
B-Viren. Dissertation der Fakultät für Biologie der Ludwig-Maximilians-<br />
Universität München, 1992.<br />
185. Fernholz, D., G. Wildner, and H. Will, Minor envelope proteins of duck hepatitis<br />
B virus are initiated at internal pre-S AUG codons but are not essential for<br />
infectivity. Virology, 1993. 197(1): p. 64-73.<br />
186. Nassal, M., Hepatitis B virus morphogenesis. Curr Top Microbiol Immunol,<br />
1996. 214: p. 297-337.<br />
187. Fricks, C.E. and J.M. Hogle, Cell-induced conformational change in poliovirus:<br />
externalization of the amino terminus of VP1 is responsible for liposome<br />
binding. J Virol, 1990. 64(5): p. 1934-45.<br />
188. Jilbert, A.R., et al., Kinetics of duck hepatitis B virus infection following low dose<br />
virus inoculation: one virus DNA genome is infectious in neonatal ducks.<br />
Virology, 1996. 226(2): p. 338-45.<br />
189. Jilbert, A.R., et al., Virus-liver cell interactions in duck hepatitis B virus infection.<br />
A study of virus dissemination within the liver. Gastroenterology, 1988. 95(5): p.<br />
1375-82.<br />
190. Cheung, R.C., et al., Epitope-specific antibody response to the surface antigen<br />
of duck hepatitis B virus in infected ducks. Virology, 1990. 176(2): p. 546-52.<br />
191. Sunyach, C., et al., Residues critical for duck hepatitis B virus neutralization are<br />
involved in host cell interaction. J Virol, 1999. 73(4): p. 2569-75.<br />
192. Johnson, D.C. and M.T. Huber, Directed egress of animal viruses promotes<br />
cell-to-cell spread. J Virol, 2002. 76(1): p. 1-8.<br />
193. Maurice, M., et al., Formation of plasma membrane domains in rat hepatocytes<br />
and hepatoma cell lines in culture. J Cell Sci, 1988. 90(Pt 1): p. 79-92.<br />
194. Zegers, M.M. and D. Hoekstra, Mechanisms and functional features of<br />
polarized membrane traffic in epithelial and hepatic cells. Biochem J, 1998.<br />
336(Pt 2): p. 257-69.<br />
195. Laemmli, U.K., Cleavage of structural proteins during the assembly of the head<br />
of bacteriophage T4. Nature, 1970. 227(259): p. 680-5.<br />
196. Sprengel, R., et al., Cloned duck hepatitis B virus DNA is infectious in Pekin<br />
ducks. J Virol, 1984. 52(3): p. 932-7.<br />
197. Saiki, R.K., et al., Enzymatic amplification of beta-globin genomic sequences<br />
and restriction site analysis for diagnosis of sickle cell anemia. Science, 1985.<br />
230(4732): p. 1350-4.<br />
198. Brown, D.A. and J.K. Rose, Sorting of GPI-anchored proteins to glycolipidenriched<br />
membrane subdomains during transport to the apical cell surface.<br />
Cell, 1992. 68(3): p. 533-44.<br />
199. Southern, E.M., Detection of specific sequences among DNA fragments<br />
separated by gel electrophoresis. J Mol Biol, 1975. 98(3): p. 503-17.<br />
153
VI. Anhang<br />
1. Abkürzungen<br />
ß-ME ß-Mercaptoethanol<br />
µm Mikrometer, 10 -6 m<br />
Abb. Abbildung<br />
AP-2 Adaptorprotein 2<br />
APS Ammoniumpersulfat<br />
AS Aminosäure<br />
ATP Adenosintriphosphat<br />
bp Basenpaare<br />
BSA bovines Serumalbumin<br />
cccDNA covalently closed circular DNA, virale DNA in kovalent<br />
CsCl Cäsiumchlorid<br />
CT Cholera-Toxin<br />
geschlossener, zirkulärer Form<br />
CTB Cholera-Toxin Untereinheit B<br />
DHBc Core-Protein von DHBV<br />
DHBe e-Antigen, early antigen, frühes Antigen, PräC<br />
DHBV duck hepatitis B virus, Enten-Hepatitis B-Virus<br />
DMSO Dimethylsulfoxid<br />
DNA deoxyribonucleic acid, Desoxyribonukleinsäure<br />
DR direct repeat, direkte Wiederholungssequenz<br />
DTT Dithiothreitol<br />
EDTA Ethylendiaminetraazetat<br />
EEA1 early endosomal antigen 1, frühes endosomales Antigen<br />
ER endoplasmatisches Retikulum<br />
FCS fetal calf serum, fötales Kälberserum<br />
FITC Fluoreszeinthiozyanat<br />
GE genome equivalents, Genomäquivalente<br />
GFP green fluorescent protein, grün fluoreszierendes Protein<br />
Anhang<br />
gp180 Glykoprotein von 180 kDa Größe, duck carboxypeptidase D,<br />
GTP Guanosintriphosphat<br />
h St<strong>und</strong>e<br />
DCPD, Carboxypeptidase D aus der Ente<br />
HBc Hepatitis B-Core-Protein<br />
HBV humanes Hepatitis B-Virus<br />
HBsAg Hepatitis B-Oberflächen-Antigen, Hepatitis B surface antigen<br />
Hepadnaviren Hepatitis-DNA-Viren<br />
154
HHBV heron HBV, Reiher-HBV<br />
HIV humanes Imm<strong>und</strong>efizienzvirus<br />
HRPO horseradish peroxidase, Peroxidase aus Meerettich<br />
HRV humanes Rhinovirus<br />
Hsc70 heat shock cognate protein 70, Hitzeschockprotein 70<br />
IgG Immunglobulin G<br />
kb Kilobasen, 1.000 bp<br />
kDa Kilodalton, 1.000 Dalton<br />
L grosses virales Oberflächenprotein<br />
LDL low density lipoprotein, Lipoprotein niedriger Dichte<br />
MßCD Methyl-ß-Cyclodextrin<br />
MGE multiplicity of GE, Genomäquivalente pro Zelle<br />
min Minuten<br />
mM millimolar, 10 -3 M<br />
MOI multiplicity of infection, Zahl der infektiösen Viren pro Zelle<br />
mRNA messenger RNA, Boten-RNA<br />
MTs Mikrotubuli<br />
NLS nuclear localization signal, nukleäres Lokalisationssignal<br />
nm Nanometer, 10 -9 m<br />
nM nanomolar, 10 -9 M<br />
ORF open reading frame, offener Leserahmen<br />
Anhang<br />
PBS phosphate buffered saline, Phosphat-gepufferte Salzlösung<br />
PCR polymerase chain reaction, Polymerase-Kettenreaktion<br />
PDHs primary duck hepatocytes, primäre Entenhepatozyten<br />
PEG Polyethylenglykol<br />
rcDNA relaxed circular DNA, virale DNA in relaxierter, zirkulärer Form<br />
RNA ribonucleic acid, Ribonukleinsäure<br />
rpm revolutions per minute, Umdrehungen pro Minute<br />
RT Raumtemperatur<br />
S kleines virales Oberflächenprotein<br />
SDS sodium dodecylsulfate, Natriumdodecylsulfat<br />
SDS-PAGE SDS-Polyacrylamid-Gelelektrophorese<br />
SNARE soluble N-ethylmaleimide-sensitive factor attachment protein<br />
receptor, löslicher N-Ethylmaleimid-sensitiver Faktor-<br />
Bindungsprotein-Rezeptor<br />
STEM surface connected tubules entering macrophages, mit der<br />
Zelloberfläche verb<strong>und</strong>ene Röhren, die in Makrophagen<br />
nachweisbar sind<br />
SVPs subvirale Partikel<br />
SV40 simian virus 40, Affenvirus 40<br />
TBS Tris buffered saline, Tris-gepufferte Salzlösung<br />
155
TCA Trichloressigsäure<br />
TP terminales Protein<br />
TRITC Tetramethylrhodamin-Isothiocyanat<br />
U Unit<br />
V Volt<br />
vATPase vesikuläre ATPase<br />
WHBV woodchuck HBV, HBV des Waldmurmeltiers<br />
WHO world health organization, Weltges<strong>und</strong>heitsorganisation<br />
wt Wildtyp<br />
Anhang<br />
156
2. Lebenslauf<br />
Geburtsdatum 05.09.1978<br />
Geburtsort Frankfurt am Main<br />
Schulausbildung<br />
1984 bis 1988 Gr<strong>und</strong>schule in Mörfelden-Walldorf<br />
1988 bis 1997 Prälat-Diehl-Gymnasium in Groß-Gerau<br />
Studium<br />
Abitur, Note 1,6<br />
Anhang<br />
1997 bis 2002 Studium der Biologie an der Technischen Universität Darmstadt<br />
2002 Diplomarbeit am Heinrich-Pette-Institut für experimentelle<br />
Praktika<br />
Immunologie <strong>und</strong> Virologie an der Universität Hamburg,<br />
Abteilung Allgemeine Virologie (Prof. H. Will)<br />
Titel: ‚Studien zu Aufnahme <strong>und</strong> intrazellulärem Transport von<br />
Hepatitis B-Viren’<br />
Diplom, Note sehr gut<br />
2000 Deutsches Krebsforschungszentrum Heidelberg, Abteilung<br />
Tumorbiochemie (Prof. D. Keppler)<br />
2001 Heinrich-Pette-Institut für experimentelle Immunologie <strong>und</strong><br />
Aktuelle Tätigkeit<br />
Virologie an der Universität Hamburg, Abteilung für Allgemeine<br />
Virologie (Prof. H. Will)<br />
Seit 2002 Wissenschaftliche Angestellte im Heinrich-Pette-Institut für<br />
experimentelle Immunologie <strong>und</strong> Virologie an der Universität<br />
Hamburg, Abteilung für Allgemeine Virologie<br />
Promotionsstudium an der Technischen Universität Darmstadt<br />
157
3. Publikationen<br />
3.1 Paper<br />
Anhang<br />
Funk, A., Mhamdi, M., Lin, L., Will, H. <strong>und</strong> Sirma, H. (2004). Itinerary of hepatitis B<br />
viruses: delineation of restriction points critical for infectious entry. J Virol.<br />
78(15): 8289-8300.<br />
Funk, A., Hohenberg, H., Mhamdi, H., Will, H. <strong>und</strong> Sirma, H. (2004) Spread of<br />
hepatitis B viruses in vitro requires extracellular progeny and may be<br />
codetermined by polarized egress. J Virol. 78(8): 3977-3983.<br />
Funk, A.*, Stöckl, L.*, Kopitzki, A., Brandenburg, B., Oess, S., Will, H., Sirma, H. <strong>und</strong><br />
Hildt, E. (2004) Identification of a novel structural motif critical for infectivity of<br />
hepatitis B viruses. Zur Veröffentlichung eingereicht. * Gleichwertige Erstautoren.<br />
Lin, L., Prassolov, A., Funk, A., Quinn, L., Hohenberg, H., Frölich, K., Newbold, J.,<br />
Ludwig, A., Will, H., Sirma, H. <strong>und</strong> Steinbach, F. (2004). Evidence from nature:<br />
interspecies spread of avian hepatitis B viruses. Zur Veröffentlichung eingereicht.<br />
3.2 Buchartikel<br />
Funk, A., Lin, L., Mhamdi, M., Will, H. <strong>und</strong> Sirma, H. (2004) Determinants of<br />
hepadnaviral species and liver cell tropism. In: M. Roggendorf (ed.), Monographs in<br />
Virology: Current models of viral hepatitis. S. Karger, Basel. Im Druck.<br />
Sirma, H., Funk, A., Hohenberg, H., Petrimpol, M., Mhamdi, M., Lin, L., Schubert, U.<br />
<strong>und</strong> Will, H. (2004) Functional modulation of virus-cell interactions by drugs: new<br />
concepts for treatment of viral hepatitis and hepatocellular carcinoma. In:<br />
Proceedings of the 11 th International Symposium on Viral Hepatitis and Liver Disease.<br />
Im Druck.<br />
158
3.3 Vorträge<br />
Anhang<br />
Funk, A., Lin, L., Will, H. <strong>und</strong> Sirma, H. (2003). Microtubules are essential for a<br />
postentry step in the early life cycle of hepatitis B viruses. Jährliche Tagung der<br />
Gesellschaft für Virologie, Berlin<br />
Funk, A. <strong>und</strong> Will, H. (2003). Infectious entry of hepatitis B viruses: delineation of<br />
restriction points critical for viral infection in vitro. The Molecular Biology of<br />
Hepatitis B and Hepatitis C Viruses. Oderbrück<br />
Funk, A., Lin, L., Will, H. <strong>und</strong> Sirma, H. (2003). Infectious entry of hepatitis B<br />
viruses: delineation of restriction points critical for viral infection in vitro. The<br />
molecular biology of hepatitis B viruses, Bergamo, Italien<br />
Mhamdi, M., Hohenberg, H., Funk, A., Lin, L., Will, H. <strong>und</strong> Sirma, H. (2003).<br />
Mechanisms regulating budding of hepatitis B viruses. The molecular biology of<br />
hepatitis B viruses, Bergamo, Italien<br />
Sirma, H., Petrimpol, M., Hohenberg, H., Funk, A., Prassolov, A., Schubert, U. <strong>und</strong> Will,<br />
H. (2003). The ubiquitin-proteasome pathway is essential for the propagation of<br />
hepadnaviruses. Jährliche Tagung der Gesellschaft für Virologie, Berlin<br />
Sirma, H., Hohenberg, H., Schneider, C., Funk, A., Meins, T. <strong>und</strong> Will, H. (2003).<br />
Ultrastructural analysis of hepadnavirus assembly and budding by use of a novel<br />
liver organoid and culture system. Jährliche Tagung der Gesellschaft für Virologie,<br />
Berlin<br />
Funk, A., Mhamdi, M., Lin, L., Will, H. <strong>und</strong> Sirma, H. (2004). Infectious itinerary of<br />
hepatitis B viruses: delineation of steps restricting viral entry. Jährliche Tagung<br />
der Gesellschaft für Virologie, Tübingen<br />
Mhamdi, M., Hohenberg, H., Funk, A., Lin, L., Will, H. <strong>und</strong> Sirma, H. (2004). Budding<br />
and secretion of hepatitis B viruses involves an endosome-like compartment.<br />
Jährliche Tagung der Gesellschaft für Virologie, Tübingen<br />
159
Anhang<br />
Funk, A., Mhamdi, M., Lin, L., Lambert, C., Prange, R., Hohenberg, H., Will, H. <strong>und</strong><br />
Sirma, H. (2004). Cholesterol is essential for the structural integrity and infectivity<br />
of hepatitis B viruses. The molecular biology of hepatitis B viruses, Woods Hole, USA<br />
Stöckl, L., Funk, A., Kopitzki, A., Will, H., Sirma, H. <strong>und</strong> Hildt, E. (2004). Identification<br />
of a novel structural motif crucial for infectivity of hepatitis B viruses. The<br />
molecular biology of hepatitis B viruses, Woods Hole, USA<br />
3.4 Poster<br />
Funk, A., Will, H. <strong>und</strong> Sirma, H. (2003). Spreading of duck hepatitis B virus mainly<br />
occurs via extracellular progeny. The molecular biology of hepatitis B viruses,<br />
Bergamo, Italien<br />
Stoeckl, L., Funk, A., Kopitzki, A., Will, H., Sirma, H. <strong>und</strong> Hildt, E. (2003). Destruction<br />
of the preS-associated cell permeability abolishes the infectivity of DHBV. The<br />
molecular biology of hepatitis B viruses, Bergamo, Italien<br />
Lin, L., Prassolov, A., Hohenberg, H., Frölich, K., Newbold, J., Funk, A., Krone, O.,<br />
Steinbach, F., Will, H. <strong>und</strong> Sirma, H. (2003, 2004). HHBV from heron species<br />
infected in nature: unusual high preS sequence conservation and spread across<br />
heron species. The molecular biology of hepatitis B viruses, Bergamo, Italien;<br />
Jährliche Tagung der Gesellschaft für Virologie, Tübingen<br />
Funk, A., Hohenberg, H., Will, H. <strong>und</strong> Sirma, H. (2004). Spread of hepatitis B viruses<br />
in vitro requires extracellular progeny and may be codetermined by polarized<br />
egress. Jährliche Tagung der Gesellschaft für Virologie, Tübingen<br />
Stöckl, L., Funk, A., Moebs, M., Kopitzki, A., Will, H., Sirma, H. <strong>und</strong> Hildt, E. (2004).<br />
Identification of a novel structural motif crucial for infectivity of hepadnavirus.<br />
Jährliche Tagung der Gesellschaft für Virologie, Tübingen<br />
Lin, L., Ishikawa, T., Huang, J., Funk, A., Will, H. <strong>und</strong> Sirma, H. (2004). Breaking the<br />
host barrier of hepatitis B viruses: requirement for a cooperative-synergistic<br />
interaction between two domains in the preS-region. The molecular biology of<br />
hepatitis B viruses, Woods Hole, USA<br />
160
Anhang<br />
Funk, A., Mhamdi, M., Hohenberg, H., Hildt, E., Will, H. <strong>und</strong> Sirma, H. (2004).<br />
Infectious entry of hepadnaviruses is mediated by endocytosis. The molecular<br />
biology of hepatitis B viruses, Woods Hole, USA<br />
161
Die vorliegende Arbeit wurde von Juli 2002 bis Oktober 2004 am<br />
Heinrich-Pette-Institut für experimentelle Immunologie <strong>und</strong> Virologie<br />
in der Abteilung für Allgemeine Virologie angefertigt.<br />
Hiermit erkläre ich an Eides statt, dass ich die vorliegende<br />
Dissertation selbstständig <strong>und</strong> nur mit den angegebenen Hilfsmitteln<br />
angefertigt habe.<br />
Hamburg, den 01.10.2004<br />
Anneke Funk














