

Instru- Zusammenfassung
Instru- Zusammenfassung
Instru- Zusammenfassung

Sie wollen auch ein ePaper? Erhöhen Sie die Reichweite Ihrer Titel.
YUMPU macht aus Druck-PDFs automatisch weboptimierte ePaper, die Google liebt.
<strong>Instru</strong>- <strong>Zusammenfassung</strong><br />
Elektrochemie<br />
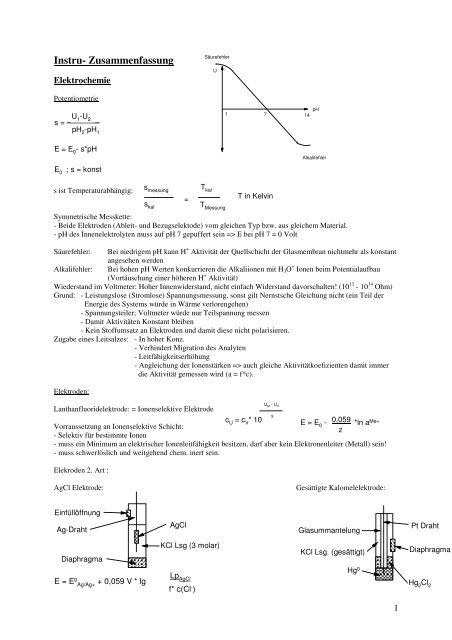
Potentiometrie<br />
s = U 1 -U 2<br />
pH 2 -pH 1<br />
E = E 0 - s*pH<br />
E 0 ; s = konst<br />
s ist Temperaturabhängig:<br />
Symmetrische Messkette:<br />
- Beide Elektroden (Ableit- und Bezugselektode) vom gleichen Typ bzw. aus gleichem Material.<br />
- pH des Innenelektrolyten muss auf pH 7 gepuffert sein => E bei pH 7 = 0 Volt<br />
Säurefehler: Bei niedrigem pH kann H + Aktivität der Quellschicht der Glasmembran nichtmehr als konstant<br />
angesehen werden<br />
Alkalifehler: Bei hohen pH Werten konkurrieren die Alkaliionen mit H3O + Ionen beim Potentialaufbau<br />
(Vortäuschung einer höheren H + Aktivität)<br />
Wiederstand im Voltmeter: Hoher Innenwiderstand, nicht einfach Widerstand davorschalten! (10 13 - 10 14 Ohm)<br />
Grund: - Leistungslose (Stromlose) Spannungsmessung, sonst gilt Nernstsche Gleichung nicht (ein Teil der<br />
Energie des Systems würde in Wärme verlorengehen)<br />
- Spannungsteiler; Voltmeter würde nur Teilspannung messen<br />
- Damit Aktivitäten Konstant bleiben<br />
- Kein Stoffumsatz an Elektroden und damit diese nicht polarisieren.<br />
Zugabe eines Leitsalzes: - In hoher Konz.<br />
- Verhindert Migration des Analyten<br />
- Leitfähigkeitserhöhung<br />
- Angleichung der Ionenstärken => auch gleiche Aktivitätkoefizienten damit immer<br />
die Aktivität gemessen wird (a = f*c).<br />
Elektroden:<br />
Lanthanfluoridelektrode: = Ionenselektive Elektrode<br />
Vorraussetzung an Ionenselektive Schicht:<br />
- Selektiv für bestimmte Ionen<br />
- muss ein Minimum an elektrischer Ionenleitfähigkeit besitzen, darf aber kein Elektronenleiter (Metall) sein!<br />
- muss schwerlöslich und weitgehend chem. inert sein.<br />
Elekroden 2. Art :<br />
AgCl Elektrode: Gesättigte Kalomelelektrode:<br />
Einfüllöffnung<br />
Ag-Draht<br />
Diaphragma<br />
E = E0 Ag/Ag+ + 0,059 V * lg<br />
s messung<br />
s kal<br />
=<br />
AgCl<br />
Lp AgCl<br />
f* c(Cl - )<br />
Säurefehler<br />
T kal<br />
U<br />
T Messung<br />
KCl Lsg (3 molar)<br />
1<br />
T in Kelvin<br />
c U = c V * 10<br />
pH<br />
7 14<br />
U M - U V<br />
s<br />
Alkalifehler<br />
E = E 0 - *ln a Me+<br />
0,059<br />
z<br />
Glasummantelung<br />
KCl Lsg. (gesättigt)<br />
Hg 0<br />
1<br />
Pt Draht<br />
Diaphragma<br />
Hg 2 Cl 2
Glaselektrode:<br />
Coulometrie<br />
galvanostatische Coulometrie potentiostatische Coulometrie<br />
- galvanostat. = konst. Stromstärke<br />
=> konst. Reaktionsgeschwindigkeit<br />
- mit Gegenelektrode<br />
- mit Zwischenreagens<br />
- Analyt nimmt linear mit der Zeit ab<br />
- mit Diaphragma<br />
- auch "Coulometrische Titration" genannt<br />
- keine Bezugselektrode<br />
Messbedingungen:<br />
- Konst Stromstärke (I)<br />
- keine Nebenreaktionen an der Arbeitselektrode<br />
=> Edukt des Zwischenreagenzes in großem<br />
Überschuss<br />
=> Zwischenreagens mit seinem Edukt ist reversibles<br />
Redox Paar<br />
- Schnelle quantitative Umsetzung des Analyten mit<br />
Zwischenreagens<br />
Formeln:<br />
Q = i * t<br />
Q = z * n * F F = 96487 C/mol<br />
n R = Q<br />
z * F<br />
m =<br />
ges. KCl Lsg<br />
Q * Mr<br />
z * F<br />
= I * t<br />
z * F<br />
AgCl<br />
Fritte<br />
Ionenselektive Glasmembran<br />
- konstantes Potential der Arbeitselektrode<br />
- mit Gegenelektrode<br />
- direkte Umsetzung des zu bestimmenden Stoffes<br />
- Unter konstanten Elektrolysebedingungen ist die<br />
Stromstärke der Konzentration des Analyten<br />
proportional. (Reaktion 1.Ordnung)<br />
Messbedingungen:<br />
- keine Nebenreaktion (100% Stromausbeute)<br />
- Wahl des geeigneten Potentials der Arbeitselektrode<br />
- Ausschluss von Sauerstoff bei der Reaktion<br />
Formeln:<br />
I t = I 0 * e -k*t<br />
Integriert:<br />
ln I t = ln I 0 - k * t<br />
Q unendlich =<br />
i 0<br />
k<br />
Abkürzen der Messung: k nur dann konstant wenn:<br />
- Potential der Arbeitselektrode konstant<br />
- Transportgeschwindigkeit der Lösung konstant<br />
(Diffusion, Konvektion)<br />
- Oberfläche der Elektrode darf sich nicht ändern<br />
2
Konduktometrie:<br />
L = κ *<br />
Λ: Leitwert (S)<br />
κ: spez. Leitfähigkeit des Leiters<br />
l: Länge des Leiters<br />
q: Querschnitt des Leiters<br />
Zellkonstante:<br />
Molare Leitfähigkeit:<br />
Leitfähigkeit hängt ab von:<br />
- Zahl der Ionen bzw. Konz. c<br />
- Anzahl an Elektronenladungen z<br />
- Wanderungsgeschwindigkeit v bzw. Ionenbeweglichkeit u<br />
Λ: molare Leitfähigkeit (S*cm 2 K =<br />
l<br />
q<br />
κ = Λ * c<br />
/mol)<br />
spez. Leitfähigkeit proportinal zur Konzentration<br />
Aber: Molare Leitfähigkeit umgekehrt proportional zur Konz.<br />
Abschwächung des auf ein Ion wirkendes elektr. Feld surch Ionen gegensätzlicher<br />
Ladung. "Bremseffekt" steigt mit wachsender Konzentration.<br />
Molare Leitfähigkeit bei unendlicher Verdünnung<br />
Kohlrausch: Λ = Λ Messaufbau: Wheatstonsche Brücke:<br />
0 - κ * c 1/2<br />
Λ<br />
q<br />
l<br />
κ =<br />
c 1/2<br />
Wechselspannung da:<br />
- keine elektr. Umsetzung<br />
- keine Polarisation der Elektroden<br />
- keine Migration der Teilchen<br />
! platinierte Elektroden !<br />
Konduktometrische Titrationskurven:<br />
L<br />
Λ =<br />
κ<br />
c<br />
τ<br />
l<br />
q<br />
Wechselspannungsquelle<br />
R1 R2<br />
Nullindikator<br />
Messlsg Regelbarer Widerstand<br />
starke Säure gegen starke Base Starke + schwache Säure gegen starke Base<br />
L<br />
τ<br />
3
Polarographe; Voltammetrie<br />
Biamperometrische Endpunktserfassung Bivoltametrische Endpunktsbestimmung<br />
= Potential bleibt konstant<br />
= Stromstärke bleibt konstant<br />
- Gemessen wird die Stromstärke in Abhängigkeit von<br />
der Reagenszugabe<br />
Amperemeter<br />
Voltmeter<br />
Gleichspannungsquelle<br />
Potentiometer zum<br />
Konstanthalten der<br />
Spannung<br />
Pt - Elektroden<br />
Es fließt nur dann ein meßbarer Strom, wenn beide<br />
Elektroden depolarisiert sind.<br />
Dies ist dann der Fall, wenn die Lösung ein<br />
reversibles Redoxpaar enthält.<br />
Stromstärke ist klein, wenn die Lsg. nur ireversible<br />
Redoxpaare enthält oder nur einen Teil reversible<br />
I<br />
I<br />
I<br />
τ<br />
τ<br />
Analyt, bzw.<br />
Titrator ist rev. Redoxpaar<br />
τ<br />
Analyt und Titrator sind<br />
reversible Redoxpare<br />
U = R * I R = U / I<br />
- Gemessen wird die Spannung in Abhängigkeit von<br />
der Reagenszugabe<br />
Widerstand<br />
Voltmeter<br />
Gleichspannungsquelle ca 10 V<br />
Pt - Elektroden<br />
Widerstand muss mind. 100 mal größer sein als der<br />
Widerstand der Lösung. R = 10 7 Ohm<br />
In Anwesenheit eines reversiblen Redoxpaaressind<br />
beide Elektroden depolarisiert => RM ist klein => U ist<br />
klein<br />
Bei Abwesenheit eines reversiblen Resoxpaares sind<br />
beide Elektroden depolarisiert => RM ist groß => U ist<br />
groß. (RM = Widerstand der Lösung)<br />
U<br />
U<br />
U<br />
reversible Redoxpare<br />
τ Analyt, bzw.<br />
τ Titrator ist rev. Redoxpaar<br />
τ Analyt und Titrator sind<br />
4
I<br />
Voltammetrie / Polaroraphie<br />
Allgemein: Polarisierte Elektrode: Eine Elektrode ist polarisiert, wenn es Potentialbereiche gibt, in<br />
denen sie Stromstärke, unabhängig vom Potential der Elektrode,<br />
sehr klein, (im Idealfall null) ist.<br />
- Potantialbereich in dem die Elektrode polarisierbar ist hängt von<br />
der Zusammensetzung der Lösung ab.<br />
- Bei einer ideal polariserten Elektrode verhält sich das System<br />
Elektrode/Lösung wie ein Kondensator und nicht wie ein<br />
Ohmscher Widerstand.<br />
Polarogramm eines Reversiblen Redoxpaares:<br />
I Anodisch<br />
I Kathodisch<br />
I<br />
Elektrode ist<br />
polarisiert<br />
A<br />
E 1/2<br />
B<br />
I D<br />
Austauschstromdichte<br />
Elektrode ist<br />
polarisiert<br />
Igr:<br />
E<br />
Diffusionsgrenzstrom (Igr ~ c) ^<br />
Quantitatives Merkmal<br />
E1/2: Halbstufenpotential, Qualitatives Merkmal<br />
bei reversiblen Redoxpaaren E 0 =E1/2 z: Anzahl der übertragenen Elektronen<br />
A: Hier bestimmt die Durchtrittsreaktion die s: 0,059 V bei reversiblen Redoxpaaren<br />
Reaktionsgeschwindigkeit => Durchtrittspolarisation s > 0,059 V bei irreversiblen<br />
B: Hier bestimmt die Diffusion die Geschwindigkeit der<br />
Durchtrittspolarisation => Konzentrationspolarisation<br />
Redoxpaaren<br />
Zersetzungsspanung: - Zur Zersetzung nötige Gegenspannung<br />
- Nach Nernst berechenbar<br />
- stimmt mit der Leerlaufspannung überein,<br />
wenn keine Überspannung vorhanden<br />
- hängt von der Konzentration des Elektrolyten ab<br />
- Durch Extrapolation der Strom - Spannungskurve erhalten<br />
Zersetzungsspannung<br />
U<br />
U<br />
lg i<br />
i gr - i<br />
Fe 2+ Fe 3+ + e -<br />
I d = k * c<br />
Ilkovic- Gleichung:<br />
k = 0,732 * z * F * D 1/2 * m 1/3 * t 1/6<br />
i<br />
lg<br />
igr - i<br />
=<br />
E 1/2<br />
E<br />
z<br />
s (E - E 1/2 )<br />
5
Konzentrationspolarisation: Spannungsänderung infolge der stofflichen Veränderung der Elektrodenumgebung.<br />
Diffusionsgrenzstrom(Igr): weitere Spannungserhöhung bewirkt keine Stromstärkeerhöhung aufgrund der<br />
Konzentrationserniedrigung infolge der elektrochemischen Umsetzung in der<br />
Elektrodenumgebung.<br />
Überspannung: Die Spannung, die zur Zersetzung eines Elektrolyten tatsächlich aufgewendet werden muss,<br />
abzüglich der theoretischen Zersetzungsspannung.<br />
Polarographische Arbeitselektroden:<br />
Vorteile Nachteile<br />
Hg - Tropfelektrode - Hohe Überspannung von H/H +<br />
=> verwendbar als Katode bis 2 V<br />
- Ständig erneuerte Elektroden-<br />
oberfläche<br />
- Keine Anreicherung der Reaktions-<br />
Edelmetalle, Pt, Au,<br />
Kohlenstoff<br />
produkte in Elektrodenumgebung<br />
- Verwendbar in positiven<br />
Potentialbereichen<br />
- Hoher Regelungsaufwand<br />
- Periodische Schwankung der<br />
Stromstärke<br />
- Anodische Auflösung von Hg in<br />
positiven Potentialbereichen<br />
- Nicht verwendbar in negativen<br />
Potentialbereichen (=> H2 Entwicklung)<br />
- Veränderung der Elektrodenoberfläche<br />
- Anreicherung der Reaktionsprodukte in<br />
Elektrodenumgebung<br />
6
Optische Methoden<br />
Refraktometrie:<br />
Brechzahl: n (λ) = c: Lichtgeschwindigkeit<br />
n: Brechzahl, dimensionslos, charakteristisch<br />
abhängig von Temperatur, Druck und Wellenlänge<br />
Relativer Brechungsindex:<br />
Messung gegen Luft statt Vakuum<br />
cVakuum cMedium n (λ) =<br />
cLuft Angabe der Molrefraktion statt n:<br />
R = M R<br />
d<br />
Snelliussches Gesetz:<br />
Mr: relative Molmasse n: Brechzahl<br />
d: relative Dichte R: Molrefraktion (l/mol<br />
β<br />
Dünn nach dicht zum Lot hin Dicht zu dünn, vom Lot weg Dicht durch dünn gleich dünn<br />
durch dicht<br />
Abbè - Refraktometer:<br />
- Grenze zwischen hell und dunkel entspricht dem Grenzwinkel der Totalreflektion<br />
- Strahlen mit größerem Einfallswinkel als dem der Totalreflektion werden total reflektiert => dunkle Zone (a)<br />
- Strahlen mit kleinerem Einfallswinkel als dem der Totalreflektion gelangen zum Okular => helle Zone (b)<br />
Lampe<br />
α<br />
Polarimetrie<br />
α<br />
n² - 1<br />
n² + 1<br />
β<br />
n 1<br />
Arten von Polarisaton: - lineare Polarisation<br />
- cirkuläre Polarisation<br />
- elliptische Polarisation<br />
Erzeugung von polarisiertem Licht Erzeugung von monochromatischem Licht<br />
- Nicolsches Prisma<br />
- Polarisator<br />
n 2<br />
α<br />
Probe Okular<br />
Lampe Polarisator Analysator<br />
b<br />
a<br />
Okular<br />
β<br />
sin 90<br />
sin β<br />
c Medium<br />
n 1<br />
n 2<br />
= n Glas<br />
n Substanz<br />
- Filter<br />
- Prisma<br />
- Gitter (Reflektionsgitter)<br />
sin α<br />
sin β<br />
sin 90 = 1<br />
= n 2<br />
n 1<br />
=> n Substanz = sin β * n Glas<br />
7
Spezifische Drehung, spezifischer Drehwert<br />
α = [α] D 20 * d * c<br />
[α] D 20 =<br />
[α] D 20: spezifischer Drehwert (°*ml/dm*g)<br />
α: Ermittelter Drehwert (°)<br />
d: Schichtdicke (dm)<br />
c: Konzentration (g/ml)<br />
In der Praxis: Feststoff:<br />
Bei Feststoffen geht die Konzentratin in g/ml ein,<br />
bei Flüssigkeiten die relative Dichte zu Wasser.<br />
Flüssigkeit:<br />
Optische Rotationsdispersion:<br />
- Bestimmung der absoluten Konfiguration<br />
- Abnahme des spez. Drehwertes bei längeren Wellenlängen<br />
- Cotton Effekt: Nulldurchgang bei λmax ,[α] D 20 bei bestimmter<br />
Wellenlänge (meist Absorptionsmaximum)<br />
FES (Flammenemissionsspektroskopie)<br />
Gründe für Abweichung von der Linearität der Eichgeraden: - niedrige Konzentration => Ionisation<br />
- zu hohe Konzentration => Eigenabsorption<br />
- Qualitativ und Quantitativ<br />
- misst Emission<br />
- wie im ersten Semester, ohne quantitativen Aspekt<br />
Gültigkeit des Lambert - Beerschen - Gesetzes wird vorrausgesetzt<br />
A = log<br />
1<br />
T<br />
= log<br />
I 0<br />
I<br />
= ε * c * d<br />
Immer Kalibrierung nötig: - Standardzumischverfahren<br />
- Kalibrierungsverfahren<br />
Standardaufbau:<br />
Hohlkathodenlampe<br />
Hohlspiegel<br />
Zerstäuber<br />
α<br />
d * c<br />
Brenngas<br />
Monchromator<br />
Detektor, Ver -<br />
Stärker, Azeige<br />
Pressluft - Substanzgemisch<br />
+<br />
[α] D 20<br />
-<br />
[α] D 20<br />
= α * 100<br />
d * ϕ 20 20<br />
α = [α] D 20 * d * c<br />
anormale ORD<br />
=<br />
=<br />
°<br />
ml<br />
Cotton Effekt<br />
Normale Opt. Rotationsdispersion<br />
λ<br />
° * ml<br />
dm * g<br />
8
Vorgänge in der Flamme:<br />
Me + + x -<br />
gelöst<br />
Verdampfen<br />
des Aerosols<br />
Bolzmann - Verteilung<br />
Me + + x- Verdampfen<br />
des Salzes<br />
fest (salzform)<br />
Unerwünschte Nebenreaktion<br />
kann nicht detektiert werden<br />
Me + + x -<br />
gasförmig<br />
Me 0<br />
gasförmig<br />
Thermische<br />
Dissoziation<br />
h * ν<br />
=> Messgröße<br />
=> Beschreibt das Verhältniss der Besetzung der unterschiedlichen Energieniveaus.<br />
N*<br />
N 0<br />
= g*<br />
g 0<br />
N* = N 0 * e<br />
Me + + e -<br />
gasförmig<br />
Me o + x 0<br />
gasförmig<br />
k: Bolzmannkonstante<br />
N: Anzahl der Atome in Grund- bzw. angeregtem Zustand<br />
g: Zahl der verfügbaren Energiezustände<br />
∆E: Anregungsenergie<br />
T: Temperatur in Kelvin<br />
Ionsation<br />
Anregung<br />
Me* o (angeregt)<br />
gasförmig<br />
Daraus ergibt sich: - Je höher die Temperatur desto größer die Anzahl der angeregten Atome<br />
- Denoch befinden sich die meisten Atome im Grundzustand<br />
FES: Temperatur muss für ein verwertbares Messergebniss konstant bleiben<br />
=> misst Emission, nur angeregte Teilchen<br />
AES: Misst nur Absorption => unangeregte Teilchen, weniger Temperaturempfindlich<br />
AES (Atomemissionsspektroskopie)<br />
Schematischer Aufbau:<br />
Zerstäuber<br />
* e<br />
-∆E<br />
k*T<br />
Hohlkathodenlampe<br />
Hohlkathodenlampe<br />
-∆E<br />
k*T<br />
Brenngas<br />
Detektor, Ver -<br />
Stärker, Azeige<br />
Monchromator<br />
(man braucht nur eine<br />
Charakteristische Wellenlänge)<br />
Pressluft - Substanzgemisch<br />
- Gefüllt mit Argon oder Neongas unter Druck<br />
- Hochspannung zwischen Anode und Kathode führt zur<br />
Ionisation des Füllgases<br />
- Beschleunigung der Ar+/Ne+ Ionen zur Kathodeund<br />
herausschlagen von Elektronen aus der Kathode<br />
=> Anregung der Atome durch Kollision mit Elektronen<br />
Atomlinien der HKL sind sehr scharf, schmal und absolut spezifisch.<br />
+<br />
-<br />
+<br />
Anode<br />
Glasschild<br />
Hohlkathode Muss<br />
gleiches Material wie Analyt sein<br />
Ar/Ne Gemisch<br />
9
Graphitrohrofen: - Weniger Probenmasse nötig<br />
- Probe verbleibt länger in Strahlengang<br />
Die AAS wird in der Gasphase durchgeführt und beruht auf der Lichtabsorption durch neutrale, nicht angeregte<br />
Atome in der Flamme.<br />
UV - Vis Spektroskopie<br />
Molekülorbitale<br />
σ - Elektronen: bindendes Elektronenpaar, Relativ hohe Anregungsenergie<br />
π - Elektonen: Dobi, Dribi: besonders in Konjgation leicht anregbar<br />
n - Elektronen: Nichtbindende Elektronen, leicht anregbar<br />
Jablonski - Termschema<br />
E<br />
s 2<br />
s 1<br />
s 0<br />
Anregung<br />
3<br />
2<br />
1<br />
0<br />
3<br />
2<br />
1<br />
0<br />
3<br />
2<br />
1<br />
0<br />
Internal Conversion<br />
T 2<br />
T 1<br />
Fluoreszenz<br />
Singulettleiter Triplettleiter<br />
Rückkehr in den Grundzustand durch:<br />
Intersystem Crossing<br />
Phosphoreszenz<br />
- Internal Conversion: Strahlungslose Inaktivierung durch Vibration oder Wärmeenergie<br />
- Intersystem Crossing: Unter Spinumkehr wechsel zu energiearmem Triplettzustand und von dort aus<br />
verzögerte Strahlungsemission => Phosphoreszenz<br />
- Fluoreszenz: Emission von Strahlung ohne Spinumkehr mit Zwischenstop in ν = 0 von s1; erst von<br />
dort zurück zu s0 unter Emission von Fluoreszenz<br />
Absorptionsbanden: - Setzen sich aus Absorptionslinien zusammen<br />
- Elektronenanregung erfolgt aus unterschiedlichen Schwingungszuständen<br />
- Wechselwirkung mit Lösungsmitteln<br />
Kalibrierung laut DAB: - Wellenlängenanzeige wird mittels einer Holmium - Perchloratlösung und einer Hg-<br />
Dampf, H2 oder D2 Lampe kalibriert.<br />
Chromophore/Definition<br />
Chromophor: - verantwortlich für Absorption<br />
- enthält π oder n Elektronen<br />
Auxochrome Gruppen: - Gruppen mit freiem Elektronenpaar,<br />
die an Chromophor gebunden sind (auxo = Hilfe)<br />
=> höhere Absorption (hyperchrom) => bathochromer Effekt<br />
(da E ~ 1/λ)<br />
Hyperchrom: - Intensitätszunahme eines Absorptionsmaximums<br />
bei konst. λ (durch Polaritätserhöhung)<br />
Hypochrom: - Intensitätsabnahme bei konst. Wellenlänge<br />
π*<br />
n oder π<br />
Intensität<br />
I<br />
hypsochrom<br />
δ∗<br />
π∗<br />
n<br />
π<br />
δ<br />
hyperchrom<br />
hypochrom<br />
bathochrom<br />
λ<br />
10
Bathochromer Effekt: - Durch anhäufung chromophorer Gruppen (z.B. konj. Dobis)Verschiebung des<br />
Absorptionsmaximums zu größeren Wellenlängen.<br />
Mit steigender Anzahl konj. Dobis dteigt HOMO an und LUMO sinkt => ∆E wird kleiner und da<br />
ν ~ ν* ~ E ~ 1/λ wird bei kleinerer Energie die Wellenlänge größer.<br />
E<br />
∆E<br />
∆E ∆E<br />
1 2 3<br />
Apparative Grundlagen<br />
Lampe<br />
Monochromator Küvette<br />
Anzahl<br />
konjugierter Dobis<br />
Detektor, Verstärker,<br />
Anzeige<br />
UV - Spektroskopie VIS - Spektroskopie<br />
Lampe Deuteriumlampe (180 - Wolframhalogenlampe<br />
350 nm)<br />
(320 - 800 nm)<br />
Küvette Quarz (Q) Glas (OS)<br />
Monochromator: - Filter (gefärbtes Glas) Detektoren: - Photomultiplier (Photonenstrom wird verstärkt)<br />
- Prismen - Photowiderstand<br />
- Reflexionsgitter - Photodiode: Aus Alkalimetall werden El -<br />
herausgeschlagen => Saugspannung zur Anode<br />
ist die Messgröße<br />
Zweistrahlphotometer kompensieren die Lösungsmittelabsorption<br />
Optische Meßgrößen und Formeln<br />
c = λ * ν c: Lichtgeschwindigkeit ν = c * ν∗ ν∗: Wellenzahl<br />
λ: Wellenlänge<br />
ν: Frequenz ν∗ = 1/λ [cm -1 ] IR!<br />
E = h * ν E: Energie<br />
h: Planksche Konstante (Naturkonstante 6,62 * 10 -34 J*s)<br />
ν: Frequenz<br />
E = (ν * c)/ λ<br />
E ∼ ν ∼ ν∗ ∼ 1/λ Energie ist proportional zur Wellenzahl, Frequenz und umgekehrt proportional zur<br />
Wellenlänge.<br />
Meßgrößen für die Lichtabsorption<br />
T = I / I0 T: Transmission I0: einfallende Intensität<br />
I: Strahlungsintensität<br />
A = log 1/T A = log I0/I A: Absorption<br />
Lambert - Beersches Gesetz<br />
A = ε * d * c A = log 1/T = log I0/I = ε * d * c<br />
11
A: Absorption Lambert - Beer gilt streng genommen nur für<br />
ε: molarer Absorptionsoeffizient (l/mol*cm) - Monochromatisches Licht<br />
d: Schichtdicke (cm) - verdünnte Lösungen (c < 10 -2 mol/l) weil Brechzahl<br />
c: Konzentration (mol/l) eines Mediums Konzentrationsabhängig ist<br />
Spezifische Absorption<br />
c = 1%ig (m/V) A 1% 1cm: spezifische Absorption (100 ml/(g*cm))<br />
d = 1 cm ε: molarer Absorptionskoeffizient (l/(mol*cm))<br />
MR: relative Molmasse (g/mol)<br />
A 1% 1cm = 10ε / MR<br />
Quadratwurzelgesetz<br />
Erlaubt Abschätzung der Anzahl der Dobis aus der Wellenlänge des Absorptionsmaximums.<br />
λmax = 134 * n 1/2 + 31 λmax: Wellenlänge des Absorptionsmaximums<br />
λ<br />
n = max - 31<br />
( )<br />
134<br />
2<br />
Polyine: λ = 171 * n 1/2 + 1<br />
n: Anzahl der konjugierten Dobis<br />
Übergangsverbote:<br />
Spin - Verbot: Verbot für den Übergang vom singulettzustand in den Triplettzustand und umgekehrt<br />
Überlappungsverbot: Verbot von Elektronenübergängen bei denen sich die Orbitale nicht genügend überlappen.<br />
Symmetrieverbot: Verbot von Elektronenübergängen zwischen Elektronenzuständen gleicher Symmetrie.<br />
Verbotene Übergänge finden mit geringer Übergangswahrscheinlichkeit, und somit mit geringer Intensität statt.<br />
(Fluoreszens, Phosphoreszens)<br />
Fluorimetrie<br />
Fluoreszens tritt nur beim Übergang vom ν = 0 von s1 in s0 Zustand auf<br />
Emittierte Strahlung ist bathochrom verschoben<br />
I (λ) ~ I0 * ε * Q * K K: Gerätekonstante<br />
ε: molarer Absorptionsoeffizient<br />
Einflüsse auf die Quantenausbeute:<br />
- pH Wert - Lösungsmittel<br />
- Konzentration - Temperatur<br />
Q: Quantenausbeute<br />
Quenching: Verringerung der Quantenausbeute durch unsaubere Lösungsmittel<br />
Fluoreszensintensität: IF = 2,3 * FF * I0 * ε * d * c<br />
Q = 2,3 * F F =<br />
Vorraussetzungen für fluorimetrische Messungen:<br />
- e sollte möglichst hoch sein<br />
- extreme Reinheitsanforderungen an das Lösungsmittel<br />
- hohe Anforderungen an die Lichtquelle (konstante und starke Lichtintensität,<br />
Hg-Dampf, Xe oder Xe-Hg Lampen, Laser)<br />
Zahl der emittierten Photonen<br />
Zahl der absorbierten Photonen<br />
12
Fluoreszens und Struktur<br />
- viele Dobis und starres Grundgerüst begünstigen Fluoreszens<br />
- Abhängig von Gegenion und eventuell pH Wert<br />
- Chelatbildung kann zu Fluoreszens führen<br />
Schematischer Aufbau: Lösungsmittelspezifische Störungen<br />
Lampe<br />
Monochromator<br />
für Anregung<br />
IR Spektroskopie<br />
Rayleigh- Streuung: angeregtes Licht wird gestreut,<br />
Wellenlänge bleibt gleich<br />
Raman Streuung: angeregtes Licht wird gestreut aber<br />
Wellenlänge ändert sich<br />
(meist bathochrom)<br />
Beide Störungen treten auch bei der doppelten<br />
Wellenlänge auf. (2. Ordnung)<br />
mittleres IR ν ∗ = 400 - 4000 ν = 1/λ Ε = (h * c)/λ c = λ ∗ ν<br />
Modell des harmonischen Oszillators<br />
ν∗ = 1<br />
2 π * c<br />
k: Federkonstante (Bindungskonstante) einfach < doppelt < dreifach<br />
M: Reduzierte Masse<br />
=> Schwingungskonstante n ~ Bindungskonstante k ~ 1/M (umgekehrt proportional zur Masse)<br />
3 N - 6 = Anzahl der Absorptionsbanden für harmonischen Oszillator (gewinkelte Moleküle)<br />
3 N - 5 = Anzahl der Absorptionasbanden für anharmonischen Oszillator (lineare Moleküle)<br />
N: Anzahl der Atome im Molekül<br />
Vorraussetzung für Energieübertragung: Kalibrierung nach DAB:<br />
- Resonanz Polystyrolfilm<br />
- Dipolmometsänderung<br />
Valenzschwingungen , Energiereicher ν<br />
Deformationsschwingungen δ<br />
Symmetrische Moleküle geben kein IR Spektrum => Raman Spektroskopie (Lichtstreuung mit Veränderung der<br />
Wellenlänge)<br />
IR Spektrometer<br />
Lampe<br />
κ<br />
( M)<br />
1/2<br />
Vergleichsprobe<br />
Probe<br />
Küvette<br />
Monochromator<br />
für Emission<br />
Detektor, Verstärker,<br />
Anzeige<br />
Monochromator<br />
Detektor<br />
M = m 1 * m 2<br />
m 1 + m 2<br />
Schreiber<br />
- Lampe: Nernststift<br />
Zirkonoxid mit Oxiden<br />
seltener Erden<br />
- Küvette: Quarz und<br />
Glas absorbieren selbst<br />
(SiO2 Gruppen)<br />
=> NaCl Fenster, KBr<br />
Pressling (Kalte<br />
Schmelze, 10 t/cm 2 )<br />
13
FT IR (Fourier Transformation)<br />
Lampe<br />
! Kein Monochromator !<br />
Fourier Transformation = Rechenschritt; Interferenzspektroskopie (Interferenz = Verstärkung und Löschung)<br />
Wichtige Wellenzahlen:<br />
C<br />
C<br />
O<br />
1200 - 1000<br />
O<br />
H<br />
2000 - 1600<br />
Massenspektroskopie<br />
3700 - 2500<br />
Terminologie<br />
Massenspektrum: Häufigkeit gegen masse/Ladugszahl - Verhältniss (m/z)<br />
Totalionenstrom: Summe aller Ströme, die von den Ionen aller m/z Werte erzeugt werden<br />
Basispeak: Der intensivste Peak wird auf 100% normiert und alles andere Relativ dazu berechnet.<br />
Molekülpeak: Entspricht dem Molekülpeak<br />
eV: 1 eV ist die Energie, die ein Elektron beim Durchwandern einer Potentialdifferenz von 1V aufnimmt.<br />
Ionisationsmethoden Ionisation durch Charakteristik der<br />
Spektren<br />
Eletronendstoßionisation 70 eV Elektronen reproduzierbare Spektren<br />
mit vielfältigen<br />
Fragmentierungen<br />
Chem. Ionisation Gasionen, Plasmaionen protonierte Molekülionen<br />
durch Gas herbeigeführt<br />
wenig Fragmentierungen<br />
Desorptionsmethoden beschleunigte Atome, durch Säure/Base oder<br />
(FD, FAD, MALDI) Ionen, Photonen<br />
(KeV,MeV)<br />
Redoxreaktionen<br />
Spraymethoden ESD Elektrische<br />
Intakte, vielfachgeladene<br />
Feldgradienten und Molekülionen, minimale<br />
thermische Energie in<br />
einem Mikrospray<br />
Fragmentierung<br />
Detektion:<br />
Probe Detektor<br />
PC<br />
Magnetfeldsektorgerät:<br />
Durch Variation der Magnetfeldstärke können<br />
die Ionen nach Massen getrennt werden<br />
Schreiber<br />
C N 2300 - 2200<br />
N H<br />
3500 - 2200 Ausnahme: Deformation 1650<br />
Deformationsschwingung normalerweise nur bis 1500<br />
m<br />
z = r2 m<br />
2 U<br />
m<br />
z<br />
* B2<br />
= konst * B2<br />
Ionenquelle<br />
Anwendung<br />
Niedrige Molmassen,<br />
strukturcharakteristische<br />
Fragestellungen<br />
Niedrige Molekülmasse<br />
Bestimung der Molmasse<br />
nichtflüchtige Proben<br />
mittlerer und hoher<br />
Masse<br />
nichtflüchtige Proben<br />
mittlerer und hoher<br />
Molekülmasse<br />
Detektor<br />
14<br />
B 0
Quadrupolgerät:<br />
Durch Modulation der Spannung können Ionen<br />
bestimmter Massen den Analysator passieren<br />
Flugzeitgeräte:<br />
Flugzeit eines Ions ist der Quadratwurzel seiner<br />
Masse proportional (TOF)<br />
E = 1/2 m * v 2 = z * e * U<br />
Auswertung<br />
- Asymptotische Abnahme => Aliphat<br />
- Fast asymptotisch mit höheren Intensitäten bei höheren Massen => verzweigter Aliphat<br />
α - Spaltung: α-Bindungen zu Heteroatomen werden bevorzugt gespalten (Heteroatom stabilisiert Ladung)<br />
O<br />
C<br />
H 3<br />
Benzylspaltung:<br />
Benzoesäurederivate : meist 195 als Basispeak<br />
Allylspaltung: Allylkation ist ein stabilisiertes Kation<br />
C<br />
H 3<br />
O<br />
CH 3<br />
OR<br />
m/z = 105<br />
Wenn 106 dann<br />
McLafferty-Umlagerung<br />
CH 3<br />
Retro-Diels-Alder: Umgekehrt zur Diels-Alder Reaktion<br />
McLafferty Umlagerung: β-Spaltung mit H-Verschiebung vom γ-H<br />
Geht bei Ketonen und Säuren<br />
DBA: Doppelbindungsäquivalente<br />
+<br />
m<br />
z<br />
= 2 e * U<br />
L 2<br />
m/z = 77<br />
DBA= 1- (Anzahl der Atome + 0,5 (Anzahl der Atome mal ihrer Bindigkeit)<br />
O<br />
-<br />
+<br />
+<br />
CH 3<br />
-<br />
CH 3<br />
+<br />
m/z = 91<br />
15
Isotopenaufspaltung:<br />
Br: durchschnittlich 79,9 => 79 Br + 81 Br ~ 1:1 Chlor: 35 Cl : 37 Cl (3:1)<br />
79 81<br />
Br<br />
NMR Spektroskopie<br />
1 : 2 : 1<br />
158 160 162<br />
Formeln und physikalische Grundlagen:<br />
Kernspinvektor: |I| = (I * (I+1)) I: Kernspinquantenzahl (13C, 1H: I = 1/2)<br />
h: Plancksche Konstante<br />
1/2 * h<br />
Durch den Kernspinvektor ergibt sich ein magnetisches Moment<br />
µ 1 = |I| * γ = γ * h * (I(I+1)) 1/2<br />
Präzessionsbewegung = Larmorbewegung<br />
Br 2 Cl<br />
Analog der Kreiselbewegung der Erdachse<br />
γ: gyromagnetisches Verhältniss<br />
(charakteristisch für eine Kernsorte)<br />
Wenn die Frequenz der eingestrahlten Energie gleich der Frequenz der Larmorbewegung ist, tritt<br />
der Resonanzfall ein.<br />
Frequenz der Larmorbewegung:<br />
Bei der Relaxation wird der Besetzungsunterschied wieder hergestellt und die eingestrahlte Energie in Form von<br />
h * n wieder frei. Die Frequenz dieser Energie entspricht der der eingestrahlten Energie und wird gemessen.<br />
Bolzmann- Verteilung: Die meisten Kerne befinden sich im Grundzustand,<br />
daher müssen sehr seltene Kerne (13C) sehr lange<br />
N* = N<br />
gemessen werden<br />
0 -∆E<br />
k*T<br />
* e<br />
Erfassbare Kerne: 1 1H, 13 6C, 15 7N, 14 7N, 19 9F, 29 14Si, 31 15P,<br />
Nicht erfassbare Kerne: 16 8O<br />
Wenn sowohl die Massenzahl als auch die Ordnungszahl gerade sind, so ist der Kern nicht magnetisch aktiv.<br />
Ansonsten sind die Kerne immer magnetisch aktiv<br />
Doppelbindungsäquivalente: DBA= 1- (Anzahl der Atome + 0,5 (Anzahl der Atome mal ihrer Bindigkeit)<br />
Tieffeld<br />
7 ppm<br />
Hochfeld<br />
0 ppm<br />
µ = γ * B 0<br />
2π<br />
3 : 1<br />
37<br />
Hohes Feld: Tiefes Feld:<br />
- niedrige Frequenz - hohe Frequenz<br />
- große Abschirmung - geringe Abschirmung<br />
- hohe Elektronendichte - geringe Elektronendichte<br />
- diamagnetische Verschiebung - paramagnetische Verschiebung<br />
35<br />
9 : 3 : 1<br />
70 72 74<br />
Cl 2<br />
16
Anisotropie: Abschirmung und Enschirmung<br />
chemische Verschiebung:<br />
Zahl der möglichen Energiezustände für eine Kernsorte:<br />
N = 2 I + 1 I: Kernspinquantenzahl<br />
1 H<br />
R<br />
H<br />
O<br />
C<br />
H 2<br />
CH 2<br />
H H<br />
Aryl - CH3<br />
Alkyl<br />
10 9 8 7 6 5 4 3 2 1 0<br />
Faustregel: Je niedriger die Elektronendichte desto größer die chemische Verschiebung.<br />
13C<br />
-COOH<br />
R<br />
H<br />
S<br />
O<br />
R R<br />
O<br />
R O R<br />
CDCl 3<br />
sp 2<br />
Aromaten<br />
O<br />
R NH 2<br />
C<br />
H 2<br />
CH 2<br />
O<br />
O-H<br />
H H<br />
RO CH 3<br />
TMS<br />
220 200 180 160 140 120 100 80 60 40 20 0 -20<br />
Chromatographische Methoden<br />
Formeln:<br />
Rf - Wert<br />
Grundlegende Vorgänge:<br />
Rf =<br />
Verteilung: Verteilung zwischen zwei flüssigen Phasen<br />
Nernstscher Veteilungskoeffizient: K =<br />
δ = ω 0 (Probe) - ω 0 (Standard)<br />
ω(Gerät)<br />
sp<br />
=> d =<br />
CDCl3 (77,0)<br />
R 3 C NO 2<br />
R 2 N CH 3<br />
Entfernung Start - Substanzfleck<br />
Entfernung Start - Lösungsmittelfront<br />
c (lipophile Phase)<br />
c (hydrophile Phase)<br />
Adsorption: Anreicherung eines Stoffes an der Oberfläche eines zweiten Stoffes<br />
=> Freundlichsche Adsorptionsisotherme<br />
ω(Kern) (Hz)<br />
ω(Gerät) (MHz)<br />
sp 3<br />
CR 3<br />
= ppm<br />
Si CR 3<br />
17
Gaschromatographie (GC)<br />
Totzeit: Zeit die vom Start bis zum ersten Lösungsmittelpeak, bzw. die Zeit, die eine Substanz, die nicht<br />
adsorbiert wird, benötigt, um die Trennsäule zu passieren. (= tm)<br />
Nettoretentionszeit: tN = tN+t - tm<br />
Retentionszeit: tN+t = tN + tm<br />
Trennstufenzahl: (Zahl der theoretischen Böden)<br />
N = 5,54 *<br />
Trennstufenhöhe:<br />
H = L<br />
N<br />
Auflösung (Trennfaktor):<br />
Rs = 1,18 *<br />
Symmetriefaktor<br />
tN+t: Gesamtretentionszeit<br />
w0,5: Halbhöhenbreite des Peaks (mittig gemessen)<br />
L: Länge der Säule<br />
N: Trennstufenzahl<br />
Auflösung von mehr als 1,5 bedeutet eine<br />
Trennung bis zur Basislinie<br />
w0,05: Peakbreite bei 1/20 der Peakhöhe<br />
d: Entfernung Lot - Aufsteigender Kurvenast bei 1/20 der Peakhöhe<br />
qualitative Bestimmung: 0,8 > As >1,5 As < 1 => leading; As > 1 tailing<br />
Van Deemter Kurve:<br />
Beschreibt die Beziehung zwischen Trägergasgeschwindigkeit<br />
und Trennleistung<br />
(bzw. Anzahl der theoretischen Böden = Trennstufenzahl)<br />
zu geringe Trägergasgeschwindigkeit => Trennung drastisch verschlechtert<br />
zu hohe Trägergasgeschwindigkeit: Trennleistung nimmt langsamer ab<br />
=> lieber zu hohe TGG als zu niedrige<br />
HPLC<br />
tN+t ( w ) 0,5<br />
2<br />
As = w 0,05<br />
2d<br />
H = A +<br />
B<br />
+ CU<br />
U<br />
t N+t2 - t N+t1<br />
w 0,5 (1) + w 0,5 (2)<br />
A: Mehrwegseffekt C: Zeitbedarf beim Massenübergang<br />
B: Längsdiffusion (von Diffusionskoeffizient der mobilen Phase abhängig)<br />
- High Pressure Liquid Chromatography<br />
- Sonderform der Säulenchromatographie bei der Druck angewandt wird<br />
- Partkelgröße bestimmt Trennleistung, je kleiner und gleichmäßiger die Partikelgröße, desto höher die<br />
Trennleistung<br />
Probeneinlaßsysteme: - Dosierschleife<br />
- Einspritzvorrichtung<br />
N<br />
Trennleistung<br />
Trägergasgeschwindigkeit v<br />
18
Säulenfüllung: Reversed Phase Säule: stationäre Phase ist unpolar<br />
Herstellung:<br />
Kieselgel<br />
Detektoren:<br />
Si OH<br />
Si OH<br />
+<br />
Cl Alkyl<br />
Si<br />
Cl Alkyl<br />
Kieselgel<br />
Si O Alkyl<br />
Si<br />
Si O Alkyl<br />
GC: Wärmeleitfähigkeitsdetektor: Trägergas (H2 oder He haben hohe Wärmeleitfähigkeit) kühlt Heizdraht<br />
=> stärkerer Strom fließt<br />
Analyten haben geringere WLF => Signal<br />
Man kann auch H2O, CO2, N2 und CS2 detektieren<br />
Flammenionisationsdetektor: Analyten werden in einer Knallgasflamme verbrannt. An Brennerdüse und<br />
Flammenspitze befindet sich eine Elektrode mit Saugspannung => es bilden sich Ionen,<br />
die Strom leiten.<br />
für organische Substanzen, nicht für H2O, CO2, N2 (brennbar, KWST)<br />
HPLC: - UV/VIS Detektor; mobile Phase darf nicht absorbieren oder verunreinigt sein<br />
- Brechzahldetektor<br />
- Leitfähigkeitsdetektor: für dissoziierende Verbindungen, Wechselspannung<br />
Ionenaustauschchromatographie:<br />
Na +<br />
Probe +<br />
SO 3 -<br />
Größenausschlußchromatographie:<br />
- Trennung beruht auf der unterschiedlichen Eindringgeschwindigkeit und -dauer von Probenmolekülen<br />
unterschiedlicher Größe in die Poren<br />
- Vom physikalischen her keine stationäre Phase vorhanden<br />
- Je kleiner das Molekül, desto tiefer dringt es ein, desto später wird es eluiert (inverser Siebeffekt)<br />
=> scheinbarer Verteilungskoeffizient:<br />
K D = V e - V 0<br />
V t - V 0<br />
Trennbedingungen:<br />
Na + Probe -<br />
Probe<br />
SO -<br />
3 +<br />
NR 3 +<br />
Ve: Elutionsvolumen des Analyten<br />
Vt: Elutionsvolumen iener total permeierenden Substanz<br />
V0: Elutionsvolumen einer nicht permeierenden Substanz<br />
Temperatur: HPLC: Kleinerer Einfluß, Temp. hoch => Retentionszeit niedrig<br />
GC: Je höher die Temperatur desto kürzer bleibt die Substanz in der Säule<br />
Peakbreite nimmt ab, Peakhöhe nimmt zu, Peakfläche bleibt gleich<br />
Elutionsmittel: HPLC: isokratisch: gleichbleibende Elutionsmittelzusammensetzung während der<br />
Chromatographie. Bei LM - Gradienten kein Brechzahldetektor benutzen.<br />
OH -<br />
OH -<br />
NR 3 +<br />
+<br />
Probe -<br />
2 HCl<br />
19
Eluotrope Reihe:<br />
Derivsatisierung:<br />
Gründe für Derivatisierungen: - geringe Flüchtigkeit<br />
- Zersetzung beim Verdampfen<br />
- hohe Polarität<br />
- Einführung von Heteroatomen, die vom Detektor spezifisch angezeigt werden<br />
mögliche Derivatisierungsreaktionen:<br />
Silylierungen<br />
Acylierungen<br />
Alkylierung<br />
Statistik:<br />
Wiederfindungsrate:<br />
Standardabweichung:<br />
Relative Standardabweichung: Varianz:<br />
srel =<br />
s<br />
* 100 %<br />
x<br />
Begriffe:<br />
Petrolether<br />
Cyclohexan<br />
Benzol<br />
Chloroform<br />
Dichlormethan<br />
Diethylether<br />
Tetrahydrofuran<br />
Aceton<br />
Acetonitril<br />
Isopropanol<br />
Methanol<br />
Wasser<br />
s =<br />
WFR =<br />
Validierung: - Nachweis der Leistungsfähigkeit einer analytischen Methode<br />
- Umfasst die Gesamtheit aller sich über Planung, Durchführung und Dokumentation<br />
erstreckender Maßnahmen.<br />
Spezifität: - Eindeutige Bestimmung einer Substanz (nur dieses Teilchen macht diesen Nachweis positiv<br />
und kein Anderes)<br />
Selektivität: - relative Spezifität<br />
- unter Umständen Kombination zweier Methoden (z.B. Gehalt per UV nur nach DC)<br />
Linearität (Linearity): => linear über Meßbereich (Range)<br />
- Bestimmung über mehrere Konzentrationsstufen<br />
- Kalibrierungsgerade (Regressionsgerade)<br />
x<br />
µ<br />
Σ(x - x i ) 2<br />
n - 1<br />
* 100%<br />
Mittelwert der Meßwerte<br />
Wahren Wert<br />
v = s 2<br />
* 100 %<br />
20
Richtigkeit (Accurancy): - Frage ob der Mittelwert der Meßwerte dem Wahren (richtigen) Wert entspricht?<br />
- Wiederfindungsrate als Maß (mindestens dreifachbestimmung auf drei verschiedenen<br />
Konzentrationsstufen)<br />
WFR =<br />
x<br />
µ<br />
* 100%<br />
Mittelwert der Meßwerte<br />
Wahren Wert<br />
- Zur Bestimmung des wahren Wertes: - Analyt bekannter Reinheit (CRS)<br />
- zweite unabhängige Methode<br />
- Ableitung aus Präzission, Linearität und Spezifität<br />
Präzission (Precision): - Übereinstimmung der Ergebnisse bei Wiederholung<br />
- Standardabweichung als Maß<br />
- Wiederholpräzission (repeatability) intra-day (Wiederholstandardabweichung)<br />
- Vergleichspräzision, auch Laborpräzision inter-day<br />
=> gleiches Labor, verschiedene Analytiker und Geräte => Vergleichsstandardabweichung<br />
Meßbereich: - Nachweisgrenze (Limit of Detection) = Qualitative Bestimmung, üblicherweise im Vergleich<br />
zum Grundrauschen bestimmt<br />
- Bestimmungsgrenze (Limit of quantifikation) = Quantitative Bestimmung => nicht aus der<br />
Regressionsgeraden<br />
Robustheit: - Anfälligkeit der Systems gegenüber Parameterveränderung<br />
(gezielt Parameter falsch einstellen und Ergabnisse vergleichen)<br />
* 100 %<br />
Empfindlichkeit: - Steigung der Kalibriergeraden im Meßbereich (Wenn bei gleichem ∆x die Steigung ∆y bei<br />
Methode 1 größer ist als bei Methode 2 so ist Methode 1 empfindlicher)<br />
Zufallsfehler Systematische Fehler<br />
- statistisch - vermeidbar<br />
- unvermeidbar - Abweichung der Meßwerte in eine Richtung<br />
- durch Wiederholungsmessung beschreibbar<br />
21

![[Inhalt] [Impressum]](https://img.yumpu.com/21481154/1/184x260/inhalt-impressum.jpg?quality=85)