Congenital Cystic Adenomatoid Malformation - SSM Cardinal ...
Congenital Cystic Adenomatoid Malformation - SSM Cardinal ...
Congenital Cystic Adenomatoid Malformation - SSM Cardinal ...

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
<strong>Congenital</strong> <strong>Cystic</strong> <strong>Adenomatoid</strong> <strong>Malformation</strong><br />
What is <strong>Congenital</strong> <strong>Cystic</strong> <strong>Adenomatoid</strong> <strong>Malformation</strong><br />
(CCAM)<br />
A CCAM is a cystic mass which forms in the lung tissue of a fetus. The<br />
mass is usually located in one lung, and it does not function as normal lung<br />
tissue. The cause of a CCAM is unknown, and it is not related to anything<br />
the mother did or did not do during the pregnancy. No cases of recurrence<br />
in a sibling have been reported. We suspect that a genetic problem causes<br />
a CCAM, as if a switch has made that part of the lung stay immature. As a<br />
result, it grows faster than normal and it forms abnormal air spaces which<br />
do not function like normal lung.<br />
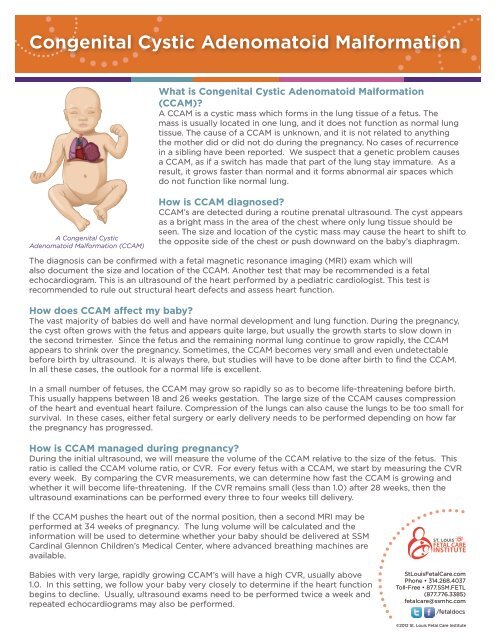
A <strong>Congenital</strong> <strong>Cystic</strong><br />
<strong>Adenomatoid</strong> <strong>Malformation</strong> (CCAM)<br />
How is CCAM diagnosed<br />
CCAM’s are detected during a routine prenatal ultrasound. The cyst appears<br />
as a bright mass in the area of the chest where only lung tissue should be<br />
seen. The size and location of the cystic mass may cause the heart to shift to<br />
the opposite side of the chest or push downward on the baby’s diaphragm.<br />
The diagnosis can be confirmed with a fetal magnetic resonance imaging (MRI) exam which will<br />
also document the size and location of the CCAM. Another test that may be recommended is a fetal<br />
echocardiogram. This is an ultrasound of the heart performed by a pediatric cardiologist. This test is<br />
recommended to rule out structural heart defects and assess heart function.<br />
How does CCAM affect my baby<br />
The vast majority of babies do well and have normal development and lung function. During the pregnancy,<br />
the cyst often grows with the fetus and appears quite large, but usually the growth starts to slow down in<br />
the second trimester. Since the fetus and the remaining normal lung continue to grow rapidly, the CCAM<br />
appears to shrink over the pregnancy. Sometimes, the CCAM becomes very small and even undetectable<br />
before birth by ultrasound. It is always there, but studies will have to be done after birth to find the CCAM.<br />
In all these cases, the outlook for a normal life is excellent.<br />
In a small number of fetuses, the CCAM may grow so rapidly so as to become life-threatening before birth.<br />
This usually happens between 18 and 26 weeks gestation. The large size of the CCAM causes compression<br />
of the heart and eventual heart failure. Compression of the lungs can also cause the lungs to be too small for<br />
survival. In these cases, either fetal surgery or early delivery needs to be performed depending on how far<br />
the pregnancy has progressed.<br />
How is CCAM managed during pregnancy<br />
During the initial ultrasound, we will measure the volume of the CCAM relative to the size of the fetus. This<br />
ratio is called the CCAM volume ratio, or CVR. For every fetus with a CCAM, we start by measuring the CVR<br />
every week. By comparing the CVR measurements, we can determine how fast the CCAM is growing and<br />
whether it will become life-threatening. If the CVR remains small (less than 1.0) after 28 weeks, then the<br />
ultrasound examinations can be performed every three to four weeks till delivery.<br />
If the CCAM pushes the heart out of the normal position, then a second MRI may be<br />
performed at 34 weeks of pregnancy. The lung volume will be calculated and the<br />
information will be used to determine whether your baby should be delivered at <strong>SSM</strong><br />
<strong>Cardinal</strong> Glennon Children’s Medical Center, where advanced breathing machines are<br />
available.<br />
Babies with very large, rapidly growing CCAM’s will have a high CVR, usually above<br />
1.0. In this setting, we follow your baby very closely to determine if the heart function<br />
begins to decline. Usually, ultrasound exams need to be performed twice a week and<br />
repeated echocardiograms may also be performed.<br />
StLouisFetalCare.com<br />
Phone • 314.268.4037<br />
Toll-Free • 877.<strong>SSM</strong>.FETL<br />
(877.776.3385)<br />
fetalcare@ssmhc.com<br />
/fetaldocs<br />
©2012 St. Louis Fetal Care Institute
<strong>Congenital</strong> <strong>Cystic</strong> <strong>Adenomatoid</strong> <strong>Malformation</strong><br />
If signs of heart failure develop, or the CVR rises to 1.6 or higher, then fetal intervention may be required.<br />
Oftentimes, prenatal steroids are the first step in the intervention. If the steroids do not stop the growth of<br />
the CCAM, then open fetal surgery to remove the mass can be a life saving option for the baby. If the CCAM<br />
has a dominant cyst of fluid then a needle can be used to drain the fluid and relieve the compression on the<br />
heart. When the CCAM is a solid mass, then open fetal surgery is necessary to remove the mass. If your<br />
baby has reached 32 weeks in pregnancy, then early delivery may be used instead of fetal surgery.<br />
How is CCAM treated after delivery<br />
If fetal intervention is not necessary the infant will be evaluated and treated after delivery. Babies with a<br />
relatively small CCAM can be born without any apparent complications. These babies typically go home and<br />
are followed as an outpatient in 2 to 4 weeks. The pediatric surgeons manage what to do with the CCAM.<br />
Often they recommend surgical removal to prevent future infection and possible malignancy. This is done<br />
though an operation in the chest and the part or lobe of the lung that contains the CCAM is removed.<br />
Babies with a moderately large CCAM’s may have some difficulty breathing after birth. Usually, these babies<br />
breathe very quickly and sometimes they require oxygen. Eating is difficult when breathing fast. These<br />
babies need to be in a neonatal intensive care unit (NICU) for stabilization and surgery is performed to<br />
remove the CCAM and to allow for the remaining normal lung to function optimally. The baby will remain in<br />
the NICU until breathing and eating improve.<br />
Rarely, a CCAM is so large that we anticipate the baby will have problems breathing right at birth. We try to<br />
predict this based on the size of the CCAM and lungs by MRI, and the degree that heart has been pushed out<br />
of the way. To avoid a crisis in the baby’s breathing at birth, we recommend that a special delivery method<br />
be used. When a baby is delivered using an EXIT procedure, the placenta and umbilical cord are maintained<br />
while we evaluate the baby’s lung function. If the baby breathes well, then the baby can be completely<br />
delivered and we can plan for removal of the CCAM later. If the baby’s breathing is compromised, then the<br />
CCAM needs to be removed immediately. The chest operation is performed while the mother’s placenta<br />
supports the baby. When the operation is finished, the cord is cut and the baby can breathe better without<br />
the compression of the CCAM.<br />
What will happen after surgery<br />
The post-operative course varies depending on when the surgery is done, the size of the CCAM, and how<br />
much lung was removed. If the CCAM is removed during the neonatal period, then commonly a breathing<br />
tube and intravenous line is needed. The baby may also have a tube in the chest to drain any fluid and help<br />
expand the lung into the chest space. The baby will not be able to eat until his or her condition has stabilized<br />
but nourishment will be given through the intravenous fluids.<br />
The baby will go home when he or she can breathe sufficiently and eat enough to maintain and gain weight.<br />
The average stay for the newborn can range from 2-3 days for small CCAM’s to 4-8 weeks for much larger<br />
ones. The long-term outcome for infants who have the cyst removed is excellent. These children usually<br />
have no limitations on their activities and have no increased risk for respiratory complications.<br />
StLouisFetalCare.com<br />
Phone • 314.268.4037<br />
Toll-Free • 877.<strong>SSM</strong>.FETL<br />
(877.776.3385)<br />
fetalcare@ssmhc.com<br />
/fetaldocs<br />
©2012 St. Louis Fetal Care Institute