Molecular-Dynamics and Monte-Carlo Simulations of Water Dynamics in CementS.-H. P. Cachia 1 , J. S. Bhatt 1 , S. V. Churakov 2 , N. C. Howlett 1 , D. A. Faux 1 , P. J. McDonald 11 Department of Physics, University of Surrey Guild<strong>for</strong>d Surrey GU2 7XH United Kingdom 2 Laboratory <strong>for</strong>Waste Management, Paul Scherrer <strong>Institute</strong>, 5232 Villigen-PSI, SwitzerlandINTRODUCTION:1 H Nuclear MagneticResonance (NMR) relaxometry has been used tocharacterise water dynamics in cement pastes onthree timescales: (i) nanosecond hopping of watermolecules across pore surfaces; (ii) microsecondresidency time <strong>for</strong> water molecules adsorbed atpore surfaces and (iii) millisecond exchange ofwater between C-S-H interlayer and gel pores. It isthe purpose of this work to seek to corroboratethese measurements using a combination ofMolecular-Dynamics (MD) and Monte-Carlo (MC)simulations. The MD is able to assess waterdynamics in nano-pores from the pico to nanosecond timescale in great detail. MC is required toextend this to longer length and timescale.M<strong>ETH</strong>ODS: Atomic trajectories of water inconfined geometries have been obtained with MDand MC simulations. These are used to calculatethe nuclear dipole-dipole correlation functions:G * (t) = 1 (3cos 2 (! 2 ! ij)"1) / (r 3 ij(t)r 3 ij(0))where the sum is over all pairs of 1 H nuclei withseparation r ij (t) and θ is the angle between thisvector at time zero and t.The correlation functions are Fourier trans<strong>for</strong>medto yield the spectral density function J(!) fromwhich the NMR spin-lattice and spin-spinrelaxation times, T 1 and T 2 respectively, arecalculated:1 = A J(!T1( 0)+ 4J(2! 0))1 = A 3J(0)+ 5J(!T2( 0)+ 2J(2! 0))where A is a constant characteristic of the materialand ! 0is the NMR frequency.RESULTS: MD simulations were per<strong>for</strong>med <strong>for</strong>bulk water, water confined between (100)-facets ofSiO 2 , Tobermorite’s basal plane and other C-S-Hanalogues.Figure 1 shows a snapshot of an MD simulation ofwater confined between two planes of SiO 2 . Theplanar spacing is d = 3.1 nm. Simulations havebeen per<strong>for</strong>med <strong>for</strong> separations as small as 1 nm.Water confined to a pore can be identified as“surface” or “bulk” dependent upon its initiallocation with respect to the SiO 2 . Figure 2 showsthe contribution to the correlation function arisingfrom bulk-bulk, surface-bulk and surface-surfaceinteractions from which the relaxation times arecalculated. There are significant differencesbetween the correlation functions of confinedwater and those <strong>for</strong> bulk water that give rise to thereduction in relaxation times. Further simulationshave followed the contribution of, <strong>for</strong> instance,surface paramagnetic impurity contributions andinter- and intra-water molecule contributions.Fig. 1: An MD snapshot of water confined between(100) SiO 2 facets. O-atoms are red, Si-atoms areyellow, H-atoms are white.Fig. 2: Total and partial G * (t) correlationfunctions. Black: total; Green: bulk-bulk; red:surface-bulk; blue: surface-surface.REFERENCES:1A. Abragam, The principle of nuclear magnetismOx<strong>for</strong>d, (1961)2 Sholl, J. Phys. C. 7, 3378 (1974)3 Faux, et al, J. Phys. C. 19, 4115 (1986)ACKNOWLEDGEMENTS:This work is supported by the European UnionSeventh Framework Programme (FP7 / 2007-2013) under grant agreement 264448 and by theU.K. Engineering and Physical Sciences ResearchCouncil, grant number EP/H033343.50
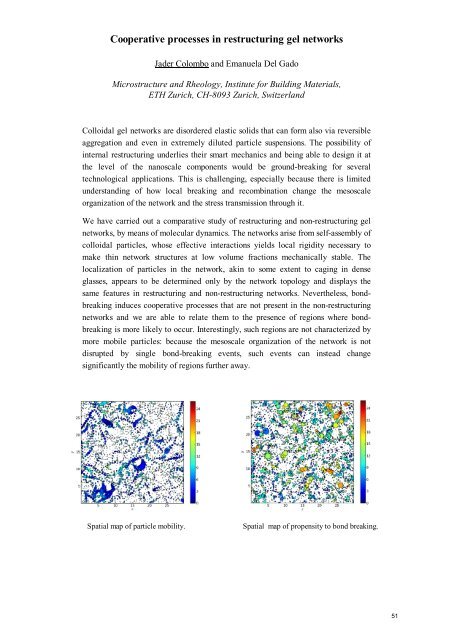
Cooperative processes in restructuring gel networksJader Colombo and Emanuela Del GadoMicrostructure and Rheology, <strong>Institute</strong> <strong>for</strong> <strong>Building</strong> <strong>Materials</strong>,<strong>ETH</strong> Zurich, CH-8093 Zurich, SwitzerlandColloidal gel networks are disordered elastic solids that can <strong>for</strong>m also via reversibleaggregation and even in extremely diluted particle suspensions. The possibility ofinternal restructuring underlies their smart mechanics and being able to design it atthe level of the nanoscale components would be ground-breaking <strong>for</strong> severaltechnological applications. This is challenging, especially because there is limitedunderstanding of how local breaking and recombination change the mesoscaleorganization of the network and the stress transmission through it.We have carried out a comparative study of restructuring and non-restructuring gelnetworks, by means of molecular dynamics. The networks arise from self-assembly ofcolloidal particles, whose effective interactions yields local rigidity necessary tomake thin network structures at low volume fractions mechanically stable. Thelocalization of particles in the network, akin to some extent to caging in denseglasses, appears to be determined only by the network topology and displays thesame features in restructuring and non-restructuring networks. Nevertheless, bondbreakinginduces cooperative processes that are not present in the non-restructuringnetworks and we are able to relate them to the presence of regions where bondbreakingis more likely to occur. Interestingly, such regions are not characterized bymore mobile particles: because the mesoscale organization of the network is notdisrupted by single bond-breaking events, such events can instead changesignificantly the mobility of regions further away.Spatial map of particle mobility.Spatial map of propensity to bond breaking.51