Saporta et al: Detecting CMT Subtypes<strong>and</strong> patients. There is little information available to guideus as to which gene to test, <strong>and</strong> testing a patient for mutationsin all commercially available CMT genes is not costeffective. Nevertheless, family planning <strong>and</strong> prognosis oftenrequire an accurate <strong>genetic</strong> diagnosis <strong>and</strong> currenttreatment trials depend on knowing the <strong>genetic</strong> cause ofa patient’s CMT even if no cures are presently available.Currently we have evaluated >1,000 patients with CMTin our clinic, many of whom have had <strong>genetic</strong> testing.We elected to analyze the results of <strong>genetic</strong> testing performedon these patients; first, to determine whether wecould analyze our phenotypic data to focus <strong>genetic</strong> testingfor patients we evaluate in the future, <strong>and</strong> second, toensure that our patient population was representative ofthose screened by various diagnostic laboratories. Wehave developed an algorithm based on clinical phenotypes,neurophysiology, <strong>and</strong> prevalence that we proposeas a guide to help focus <strong>genetic</strong> testing for various formsof CMT.Patients <strong>and</strong> MethodsCharacterization of CMT SubtypesWe included all patients evaluated at our CMT clinic between1997 <strong>and</strong> 2009. Patients were considered to have CMT if theyhad a sensorimotor peripheral neuropathy <strong>and</strong> a family historyof a similar condition. Patients without a family history of neuropathywere included if their medical history, neurophysiologicaltesting, <strong>and</strong> neurological examination were typical forCMT1, CMT2, CMTX, or CMT4. Patients were excluded ifthere were known diagnoses of acquired neuropathy includingtoxic (eg, medication-related neuropathies); metabolic (eg, diabetic),immune mediated or inflammatory (AIDP or CIDP)polyneuropathies; neuropathy related to leukodystrophy, or congenitalmuscular dystrophy; <strong>and</strong> patients with severe generalmedical conditions. First-degree or second-degree relatives of<strong>genetic</strong>ally defined patients with a CMT phenotype wereassumed to have the same mutation. Patients without an identified<strong>genetic</strong> cause were classified based on nerve conductionvelocities, physical examination, <strong>and</strong> family history.This study was approved by the Institutional ReviewBoard (IRB) at Wayne State University.Genetic Testing Hit RatesData was collected on patients for whom commercial <strong>genetic</strong>testing was ordered by Wayne State University between 2005<strong>and</strong> 2009. Hit rates were defined as the number of positiveresults for a particular gene, out of the total number of times<strong>genetic</strong> testing was ordered for that gene. As our experiencewith different <strong>subtypes</strong> of CMT has grown we have incorporatedphenotypic characteristics in our decision-making for<strong>genetic</strong> testing. Our criteria for which genes to test has evolvedover the years <strong>and</strong> our ‘‘hit rates’’ should be interpreted withthis in mind. Patients who had previously obtained positiveTABLE 1: CMT Subtype DistributionCMTSubtypenPatients withGenetically-Defined CMT(n 5 527) (%)All Patientswith CMT(n 5 787)(%)CMT1A 290 55.0 36.9CMT1B 45 8.5 5.7CMT1X 80 15.2 10.2Males 44 8.4 5.6Females 36 6.8 4.6CMT2A 21 4.0 2.7HNPP 48 9.1 6.1Total 484 91.8 61.5CMT ¼ <strong>Charcot</strong>-<strong>Marie</strong>-<strong>Tooth</strong> <strong>disease</strong>; HNPP ¼ hereditaryneuropathy with liability to pressure palsies.<strong>genetic</strong> testing for their type of CMT were not included in thehit rate analysis but were included in our phenotypic analysis.ResultsDistribution of CMT SubtypesA total of 1,024 patients were evaluated at our CMTclinic between 1997 <strong>and</strong> 2009, of which 787 were diagnosedwith CMT. Of the 237 patients who did not haveCMT, 118 were diagnosed with a different conditionwhile 119 were determined to be an unaffected familymember of a patient with CMT.Of the 787 patients with CMT (67%), 527patients had or received a specific <strong>genetic</strong> diagnosis, whilein 260 patients with CMT no specific mutation wasidentified. The most prominent CMT <strong>subtypes</strong> identifiedin our clinic were CMT1A, CMT1X, hereditary neuropathywith liability to pressure palsies (HNPP), CMT1B,<strong>and</strong> CMT2A (Table 1). All other CMT <strong>subtypes</strong>accounted for less than 1% of all patients with <strong>genetic</strong>allydefined CMT each. Only 1.8% of patients withCMT1 were without a <strong>genetic</strong> diagnosis. These patientswere defined as having a demyelinating phenotype <strong>and</strong> adominant family history. Of patients with CMT2,65.6% were without a <strong>genetic</strong> diagnosis. These patientswere defined as having an axonal phenotype <strong>and</strong> a dominantfamily history (Table 2). The distribution of <strong>genetic</strong><strong>subtypes</strong> identified in our clinic was similar to the distributionof patients identified by multiple laboratories thatperform diagnostic testing for CMT (reviewed in Engl<strong>and</strong><strong>and</strong> colleagues 7,8 ). Comparing our results to thosefrom these laboratories (their results follow in parentheses),we identified CMT1A in 82% (80%) of patientsJanuary 2011 23
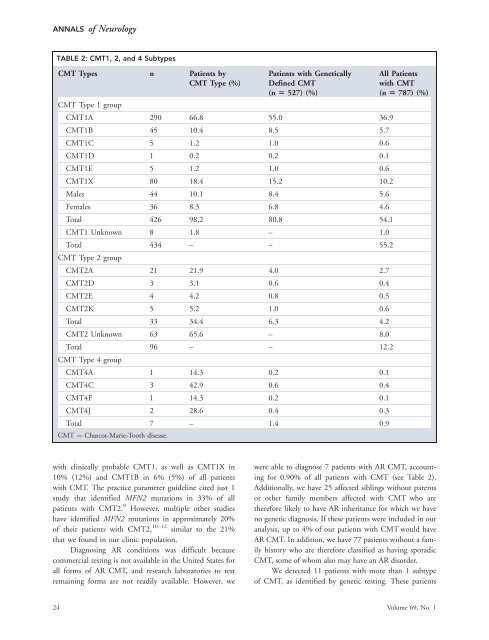
ANNALS of NeurologyTABLE 2: CMT1, 2, <strong>and</strong> 4 SubtypesCMT Types n Patients byCMT Type (%)Patients with GeneticallyDefined CMT(n 5 527) (%)CMT Type 1 groupCMT1A 290 66.8 55.0 36.9CMT1B 45 10.4 8.5 5.7CMT1C 5 1.2 1.0 0.6CMT1D 1 0.2 0.2 0.1CMT1E 5 1.2 1.0 0.6CMT1X 80 18.4 15.2 10.2Males 44 10.1 8.4 5.6Females 36 8.3 6.8 4.6Total 426 98.2 80.8 54.1CMT1 Unknown 8 1.8 – 1.0Total 434 – – 55.2CMT Type 2 groupCMT2A 21 21.9 4.0 2.7CMT2D 3 3.1 0.6 0.4CMT2E 4 4.2 0.8 0.5CMT2K 5 5.2 1.0 0.6Total 33 34.4 6.3 4.2CMT2 Unknown 63 65.6 – 8.0Total 96 – – 12.2CMT Type 4 groupCMT4A 1 14.3 0.2 0.1CMT4C 3 42.9 0.6 0.4CMT4F 1 14.3 0.2 0.1CMT4J 2 28.6 0.4 0.3Total 7 – 1.4 0.9CMT ¼ <strong>Charcot</strong>-<strong>Marie</strong>-<strong>Tooth</strong> <strong>disease</strong>.All Patientswith CMT(n 5 787) (%)with clinically probable CMT1, as well as CMT1X in10% (12%) <strong>and</strong> CMT1B in 6% (5%) of all patientswith CMT. The practice parameter guideline cited just 1study that identified MFN2 mutations in 33% of allpatients with CMT2. 9 However, multiple other studieshave identified MFN2 mutations in approximately 20%of their patients with CMT2, 10–12 similar to the 21%that we found in our clinic population.Diagnosing AR conditions was difficult becausecommercial testing is not available in the United States forall forms of AR CMT, <strong>and</strong> research laboratories to testremaining forms are not readily available. However, wewere able to diagnose 7 patients with AR CMT, accountingfor 0.90% of all patients with CMT (see Table 2).Additionally, we have 25 affected siblings without parentsor other family members affected with CMT who aretherefore likely to have AR inheritance for which we haveno <strong>genetic</strong> diagnosis. If these patients were included in ouranalysis, up to 4% of our patients with CMT would haveAR CMT. In addition, we have 77 patients without a familyhistory who are therefore classified as having sporadicCMT, some of whom also may have an AR disorder.We detected 11 patients with more than 1 subtypeof CMT, as identified by <strong>genetic</strong> testing. These patients24 Volume 69, No. 1