

Capitolo 85 Disturbi del metabolismo degli amminoacidi - Doctor33
Capitolo 85 Disturbi del metabolismo degli amminoacidi - Doctor33
Capitolo 85 Disturbi del metabolismo degli amminoacidi - Doctor33

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
566 ■ PARTE X ■ Malattie metaboliche<br />
Aciduria 3-metilglutaconica di tipo I (defi cit di 3-metilglutaconil<br />
CoA idratasi ) (vedi Fig. <strong>85</strong>-4). Questo raro disturbo autosomico<br />
recessivo si manifesta con ritardo <strong>del</strong> linguaggio, movimenti coreoatetoidi,<br />
atrofi a ottica, lieve ritardo psicomotorio e sviluppo<br />
di acidosi metabolica durante uno stato catabolico. Sono<br />
stati segnalati casi di adulti asintomatici. I pazienti eliminano<br />
attraverso le urine grandi quantità di acido 3-metilglutaconico e<br />
moderate quantità di acido 3-idrossivalerico e 3-metilglutarico.<br />
Il defi cit di 3-metilglutaconil CoA idratasi è stato evidenziato<br />
anche su colture di fi broblasti e linfoblasti. È stato suggerito il<br />
trattamento con una dieta a ridotto contenuto proteico, ma i<br />
suoi benefi ci terapeutici sul corso clinico <strong>del</strong>la malattia restano<br />
da dimostrare. La somministrazione di L-carnitina si è rivelata<br />
effi cace in un paziente. Il gene per l’enzima (AUH) è localizzato<br />
sul cromosoma 9.<br />
Aciduria 3-metilglutaconica di tipo II ( cardiomiopatia legata all’X,<br />
neutropenia, ritardo <strong>del</strong>la crescita, e aciduria 3-metilglutaconica con<br />
3-metilglutaconil CoA idratasi, sindrome di Barth ). Le manifestazioni<br />
cliniche di questo disturbo, che solitamente emerge poco dopo<br />
la nascita, includono cardiomiopatia dilatativa (che si manifesta<br />
con distress respiratorio e insuffi cienza cardiaca), ipotonia, ritardo<br />
<strong>del</strong>la crescita e neutropenia da grave a moderata. In alcuni<br />
pazienti è stata segnalata una lieve aciduria lattica e/o ipoglicemia.<br />
Se il paziente sopravvive all’infanzia, può evidenziarsi un<br />
miglioramento relativo dei sintomi nel corso <strong>del</strong>la crescita. Lo<br />
sviluppo cognitivo è solitamente normale, malgrado il ritardo<br />
<strong>del</strong>la funzione motoria.<br />
I reperti di laboratorio includono un aumento da lieve a moderato<br />
<strong>del</strong>l’escrezione urinaria <strong>degli</strong> acidi 3-metilglutaconico,<br />
Acetil-CoA + Acetil-CoA<br />
Deficit<br />
di tiolasi<br />
4<br />
CoASH<br />
Acetoacetil-CoA<br />
3-idrossi-3-metilglutaril-CoA<br />
(HMG-CoA)<br />
Acido mevalonico<br />
5<br />
Aciduria<br />
mevalonica<br />
Acido 5-fosfomevalonico<br />
Colesterolo<br />
Acetil-CoA<br />
Amminoacidi<br />
chetogenici<br />
Acidi grassi<br />
via<br />
<strong>del</strong> citrato Acetil CoA + Acetil CoA<br />
Citosol<br />
Fegato<br />
1<br />
CoASH<br />
Acetoacetil-CoA<br />
Acetil-CoA<br />
2<br />
3-idrossi-3-metilglutaril-CoA<br />
(HMG-CoA)<br />
CoA<br />
Acetoacetato<br />
Acetone 3-indrossibutirato<br />
Polmoni<br />
3<br />
Membrana mitocondriale<br />
3-metilglutarico e 2-etilidracrilico. La neutropenia è un reperto<br />
comune. In alcuni pazienti è stata segnalata la presenza di acidosi<br />
lattica, ipoglicemia e anomalie <strong>del</strong>la ultrastruttura mitocondriale.<br />
Contrariamente alla aciduria 3-metilglutaconica di tipo I, l’escrezione<br />
urinaria di acido 3-idrossiisovalerico non risulta elevata. La<br />
cardiolipina totale e le sue diverse sottoclassi sono molto ridotte<br />
in colture di fi broblasti <strong>del</strong> derma. Questi dati contribuiscono alla<br />
formulazione <strong>del</strong>la diagnosi. Il disturbo è ereditato con modalità<br />
autosomica recessiva. Il gene si trova sul cromosoma Xq28;<br />
sono state identifi cate diverse mutazioni patogenetiche. L’attività<br />
<strong>del</strong>l’enzima 3-metilglutaconil CoA idratasi è normale. La ragione<br />
<strong>del</strong>l’aumentata escrezione <strong>degli</strong> acidi organici citati non è nota,<br />
né risulta disponibile alcun trattamento effi cace.<br />
Aciduria 3-metilglutaconica di tipo III ( sindrome <strong>del</strong>l’atrofi a ottica di<br />
Costeff). Le manifestazioni cliniche includono una precoce atrofi a<br />
ottica e il successivo sviluppo di movimenti coreoatetoidi, spasticità,<br />
atassia, disartria e lieve ritardo <strong>del</strong>lo sviluppo. Tutti i pazienti<br />
segnalati (eccetto uno) sono ebrei di origine irachena abitanti<br />
in Israele. Questi soggetti eliminano moderate quantità di acido<br />
3-metilglutaconico e 3-metilglutarico. Come nella forma di tipo<br />
II, la ragione <strong>del</strong>l’aumentata escrezione di questi acidi organici<br />
non è nota. L’attività <strong>del</strong>l’enzima 3-metilglutaconil CoA idratasi<br />
risulta normale. Il disturbo è ereditato con modalità autosomica<br />
recessiva. Il gene responsabile (OPA3) si trova sul cromosoma<br />
19q13.2-13.3. Non è disponibile alcun trattamento effi cace.<br />
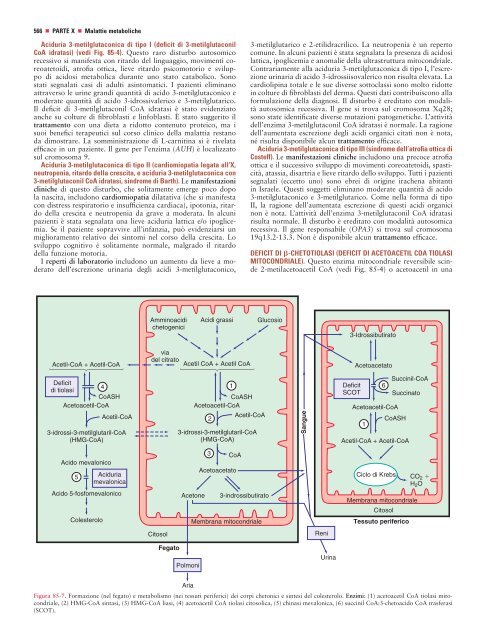
DEFICIT DI -CHETOTIOLASI (DEFICIT DI ACETOACETIL COA TIOLASI<br />
MITOCONDRIALE ). Questo enzima mitocondriale reversibile scinde<br />
2-metilacetoacetil CoA (vedi Fig. <strong>85</strong>-4) o acetoacetil in una<br />
Glucosio<br />
Acetoacetil-CoA<br />
CoASH<br />
1<br />
Aria<br />
Figura <strong>85</strong>-7. Formazione (nel fegato) e <strong>metabolismo</strong> (nei tessuti periferici) dei corpi chetonici e sintesi <strong>del</strong> colesterolo . Enzimi: (1) acetoacetil CoA tiolasi mitocondriale,<br />
(2) HMG-CoA sintasi, (3) HMG-CoA liasi, (4) acetoacetil CoA tiolasi citosolica, (5) chinasi mevalonica, (6) succinil CoA:3-chetoacido CoA trasferasi<br />
(SCOT).<br />
Sangue<br />
Reni<br />
Urina<br />
3-Idrossibutirato<br />
Deficit<br />
SCOT<br />
Acetoacetato<br />
6<br />
Acetil-CoA + Acetil-CoA<br />
Ciclo di Krebs<br />
Succinil-CoA<br />
Succinato<br />
Membrana mitocondriale<br />
Citosol<br />
Tessuto periferico<br />
CO 2 <br />
H2O




