CHAPTER 4. THERMODYNAMICS: THE FIRST LAW
CHAPTER 4. THERMODYNAMICS: THE FIRST LAW
CHAPTER 4. THERMODYNAMICS: THE FIRST LAW

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
4-1<br />
<strong>CHAPTER</strong> <strong>4.</strong> <strong><strong>THE</strong>RMODYNAMICS</strong>: <strong>THE</strong> <strong>FIRST</strong> <strong>LAW</strong><br />
A. Introduction<br />
Thermodynamics deals with the macroscopic observable properties of matter. It is a subject<br />
that is broadly applicable to all energetic processes involving large aggregates of atoms and<br />
molecules.<br />
Thermodynamics is founded upon three fundamental laws of nature. The first of these laws<br />
is the familiar law of conservation of energy, which in general terms states that the energy of the<br />
universe is constant. As with other natural laws, the validity and generality of the laws of<br />
thermodynamics have been firmly established by experimental observation. One of the major<br />
strengths of thermodynamics is that it does not rely on any assumptions regarding the fundamental<br />
structure of matter.<br />
Thermodynamics plays a major role in chemistry. It provides the theoretical basis for<br />
equilibrium as well as for most discussions of the energetics of chemical reactions. One of the<br />
primary applications of thermodynamics to chemistry is in providing the criteria for predicting<br />
whether a particular chemical reaction will proceed spontaneously.<br />
In the study of thermodynamics, it is necessary to divide the universe into the particular<br />
region in which we are interested, called the system, and everything else, called the surroundings.<br />
Usually, the system is separated from the surroundings by some kind of physical boundary. If this<br />
boundary is such that both matter and energy may be transferred between the system and the<br />
surroundings, the system is called an open system. If the boundary prevents the transfer of matter,<br />
but allows the transfer of energy, the system is called a closed system. A system for which neither<br />
the transfer of matter or energy is possible is called an isolated system. For example, a thermos<br />
bottle with the top screwed on is an isolated system because it is thermally insulated from the<br />
surroundings.<br />
B. State functions<br />
In order to specify the macroscopic state of a system, it is necessary to know the values of<br />
various parameters called state functions, such as temperature, pressure, volume, etc. When a<br />
system changes from one state to another, the final values of the state functions do not depend on
4-2<br />
how the change of state was carried out. Therefore, the changes in the values of the state functions<br />
are determined only by the initial and final states of the system. For example, consider the four states<br />
of a system composed of an ideal gas, as depicted in Fig. 4-1.<br />
path 1<br />
State 2<br />
P 2<br />
= 2 atm<br />
V 2<br />
= 22 L<br />
T 2<br />
= 546 K<br />
State 1<br />
P 1<br />
= 1 atm<br />
V 1<br />
= 22 L<br />
T 1<br />
= 273 K<br />
path 3<br />
State 4<br />
P 4<br />
= 2 atm<br />
V 4<br />
= 11 L<br />
T 4<br />
= 273 K<br />
path 2<br />
State 3<br />
P 3<br />
= 1 atm<br />
V 3<br />
= 11 L<br />
T 3<br />
= 136 K<br />
Figure 4-1. Transformation of a system from state 1 to state 4 via three different paths.<br />
Three different paths lead from state 1 to state <strong>4.</strong> However, the changes in all three of the state<br />
functions listed (P, V, and T) are the same for each path. Going from state 1 to state 4 via path 1,<br />
path 2, or path 3 results in P = 1 atm, V = 11 L, and T = 0 K. In other words, the final value<br />
of a state function does not depend on the path taken between the initial and final state. On<br />
the other hand, suppose that instead of different states of a system, the above (state) labels<br />
represented different cities on a map and the three paths represented different routes between them.<br />
It is obvious that distance is not a state function since the distance between city 1 and city 4 depends<br />
on the route taken. In the city analogy, is altitude a state function? Of course distance is not a<br />
thermodynamic variable, but two very important thermodynamic variables that are not state<br />
functions are work and heat.<br />
C. Work and heat
is<br />
=<br />
4-3<br />
Mechanical work is defined as a force applied in the direction of motion times the distance<br />
moved (displacement). Specifically,<br />
dw ' PF@ Pds ' Fdscosθ<br />
,<br />
where θ<br />
the angle between the force vector and the displacement vector. An example of the<br />
application of this formula is the calculation of the work done in raising a mass m to a height h<br />
against the force of gravity:<br />
F = ma = mg<br />
dw = Fds cos θ<br />
mgds<br />
w '<br />
h<br />
0<br />
mgds ' mgh.<br />
A particularly important form of work in chemical thermodynamics is the work associated<br />
with a pressure-volume change. The diagram in Fig. 4-2 shows a cross sectional view of a piston<br />
undergoing a compression.<br />
Figure 4-2. A piston system undergoing a compression.<br />
The applied force can be expressed as an external pressure times the cross sectional area of the<br />
piston; F = P A. Therefore,<br />
ex<br />
but<br />
dw = Fds = P Ads, ex
4-4<br />
and so<br />
Ads = dV,<br />
dw = P dV. ex<br />
The thermodynamic sign convention is that work done on the system is positive. To conform to this<br />
convention, the above expression for P-V work must include a negative sign since, work was done<br />
on the system, but because V < V , dV was negative. Therefore, the general expression for<br />
2 1<br />
differential P/V work is<br />
.<br />
If an expansion (compression) is carried out with a constant external pressure, then the work<br />
done by (on) the system is<br />
V 2<br />
dw ' !<br />
V 1<br />
P ex<br />
dV ' !P ex<br />
(V 2<br />
! V 1<br />
) ' !P ex<br />
V<br />
.<br />
Another way of performing a compression would be to keep the temperature constant and make the<br />
external pressure infinitesimally greater than the internal pressure throughout the process. This is<br />
called an isothermal reversible compression. The term reversible is derived from the fact that the<br />
process could be reversed by applying an infinitesimal change in the opposite direction. Assuming<br />
ideal behavior of the gas inside the system, the external pressure in this case is<br />
P = P = nRT/V,<br />
ex<br />
(i.e., neglecting the infinitesimal difference, the external pressure is equal to the internal pressure for<br />
a reversible compression or expansion). Therefore, the work done on the system is<br />
dw ' !<br />
V 2<br />
V 1<br />
P ex<br />
dV ' !nRT<br />
V 2<br />
dV<br />
V ' !nRTln V 2<br />
V<br />
V 1<br />
1<br />
.<br />
P-V diagrams are useful for analyzing processes involving P-V work. A P-V diagram for two
expansion processes which start at P , V and end at P , V are shown in Fig. 4-3.<br />
1 1 2 2<br />
4-5<br />
P 1<br />
a<br />
path 1<br />
b<br />
dw = -PdV<br />
P<br />
path 2<br />
c<br />
P 2<br />
V 1 V<br />
V 2<br />
Figure 4-3.<br />
A P-V diagram for two expansion processes between the same initial and final values<br />
of P and V.<br />
Path 1 consists of a constant pressure expansion from point a to point b followed by a reduction in<br />
pressure at constant volume from point b to point c. Path 2 is an isothermal reversible expansion (P in<br />
= P ) from point a to point c (shown by the hyperbolic curve or isotherm). As indicated by the<br />
ex<br />
shaded region labeled P dV, the differential work is represented by the area of a rectangle having<br />
1<br />
length P and width dV. Therefore, the total work associated with path 1 is equal to the area of the<br />
rectangle having length P and width V ! V , while the work associated with path 2 is equal to the<br />
1 2 1<br />
area under the hyperbolic curve. Clearly these two areas are not the same even though both<br />
processes involved the same changes in pressure and volume. One must conclude, therefore, that<br />
work is not a state function.<br />
Another way in which energy can be exchanged between systems is by heat flow. A system<br />
at a higher temperature may exchange energy with a system at a lower temperature, thereby lowering<br />
its own temperature and raising the temperature of the other system in the process. Since the same<br />
change in state can be achieved by either heat or work, one must conclude that like work, heat is not<br />
a state function.
4-6<br />
Exercise:<br />
Calculate the work for each of the following constant temperature processes;<br />
a). Expansion due to a sudden pressure change from P ex to P ex/<strong>4.</strong><br />
b). Expansion in two steps- from P to P /2, then from P /2 to P /<strong>4.</strong><br />
ex ex ex ex<br />
c). Reversible expansion from P ex to P ex/<strong>4.</strong><br />
What do you conclude about a reversible expansion?<br />
a). w = ! P V; V = V (P /P ) = 4V so V = 3V and<br />
2 1 1 2 1 1<br />
w = ! (P /4)(3V ) = !0.75P V .<br />
1 1 1 1<br />
b). w = ! 0.5P V = !0.5P V (V = 2V )<br />
1 1 1 1 2 1<br />
w = ! 0.25P V = !0.5P V (V = 4V )<br />
2 1 1 1 2 1<br />
w = w + w = ! P V .<br />
1 2 1 1<br />
c). w = ! nRT ln (V /V ) = ! P V ln 4 = !1.39P V .<br />
2 1 1 1 1 1<br />
As can be seen from this exercise, the reversible work is the maximum possible work<br />
that can be done by the system or the surroundings in a given process.<br />
D. The first law of thermodynamics<br />
Since heat added to a system must result in a change of the internal energy content of the<br />
system, or it must produce work, or both, one is led to expect that these quantities must be intimately<br />
related. The first law of thermodynamics expresses the fact that (a) heat and/or work are involved<br />
in the transfer of internal energy from one system to another, and (b) the total internal energy of a<br />
system must be conserved;<br />
The first Law.<br />
In the first law equation above, the quantity U represents the internal energy, which consists of the<br />
kinetic and potential energies of all the atoms and molecules of the system. Unlike heat (q) and work<br />
(w), internal energy is a state function. If this were not true, it would be possible to construct a<br />
cyclic process going from state a to state b that released U and then returned back to state a using<br />
1<br />
U , such that | U | < | U |. This is the condition for a perpetual motion machine. It is important<br />
2 2 1<br />
to understand the thermodynamic sign conventions as they apply to the first law. Namely,
4-7<br />
when heat is added to a system, q is positive,<br />
when work is done on a system, w is positive.<br />
According to the first law, when heat is transferred to or from a closed system in a process<br />
that does no work, the heat involved is equal to the internal energy change. Specifically, for a<br />
constant volume process<br />
where<br />
but<br />
and so<br />
dU = dq + dw,<br />
dw = ! P dV, ext<br />
dV = 0<br />
at constant volume.<br />
In general, when only P-V work is involved,<br />
dq = dU + P dV. ext<br />
Therefore, it is useful to define a new quantity (H), called enthalpy, such that<br />
,<br />
where P is the pressure of the system. Since U, P, and V are all state functions, H is a state function<br />
also. A differential change in H may be expressed as<br />
When P is constant,<br />
dH = dU + PdV + VdP.<br />
dH = dU + PdV,<br />
and if P = P (i.e., when the system is in mechanical equilibrium with the surroundings),<br />
ext
4-8<br />
Therefore,<br />
dH = dU + P dV. ext<br />
at constant pressure.<br />
For liquids and solids, (PV) is usually very small (typically - 0.1% of U), and hence H<br />
. U. For ideal gases,<br />
H = U + PV = U + nRT,<br />
so<br />
H = U + nRT<br />
at constant temperature. It should be noted that n is the change in the number of moles of gas.<br />
Exercise:<br />
A total of 885.7 kJ of heat is evolved in the reaction<br />
CH + 2O ! CO + 2H O<br />
4(g) 2(g) 2(g) 2 (l)<br />
o<br />
Carried out at fixed volume and 25 C. Calculate U and H for this reaction.<br />
U = q = !885.7 kJ<br />
H = U + (PV) = U + nRT; n = 1 ! 3 = !2<br />
!3<br />
H = !885.7 ! 2(8.314)(298)x10 = !890.6 kJ<br />
Exercise:<br />
A 10.0 g sample of benzene at its boiling temperature (353.2 K) is placed in contact<br />
with a resistance heater. It evaporates when 3942 J of heat have been produced by<br />
the heater. Calculate the molar enthalpy change and the the molar internal energy<br />
change. (Neglect the volume of the liquid).<br />
This is a constant pressure process in which H = U + (PV) = q.<br />
n = 10.0 g C H x 1 mol C H /78 g C H = 0.128 mol<br />
6 6 6 6 6 6<br />
H = 3942 J/0.128 mol = 30797 J/mol = 30.8 kJ/mol<br />
m<br />
U = H ! (PV ! PV )/n = H ! RT<br />
m m gas liq m
4-9<br />
!3<br />
= 30.8 kJ/mol - (8.314x10 kJ/mol-K)(353.2 K) = 27.9 kJ/mol.<br />
E. Heat capacity<br />
The amount of heat released or absorbed in a process may be determined by measuring the<br />
change in temperature of a mass in thermal contact with the process. The heat capacity is defined<br />
as the change in heat divided by the change in temperature, C = dq/dT. However, this definition is<br />
not very useful unless a path is specified, since the value of dq depends on how the process is carried<br />
out. In the laboratory, there are two convenient paths; the constant volume path, where a reaction<br />
is carried out in a closed container so that the volume cannot change, and the constant pressure path,<br />
where a reaction is carried out in an open container so that the pressure remains constant at the<br />
pressure of the surrounding atmosphere. Consider a process carried out at constant volume. Then<br />
dw = 0 and so dq = dU, therefore, the heat capacity (per mole) at constant volume is<br />
and<br />
,<br />
.<br />
For a process carried out at constant pressure, dq = dH, therefore, the heat capacity (per mole) at<br />
constant pressure is<br />
,<br />
and<br />
.<br />
Physically, the difference between C V and C P is related to the fact that when the volume is
4-10<br />
held constant, all of the heat goes into producing a corresponding change in temperature (i.e., it all<br />
goes into changing the internal energy), whereas when the pressure is held constant, some of the heat<br />
goes into work of expansion or contraction. Therefore, it is to be expected that<br />
since a given q results in a smaller T.<br />
P<br />
C P > CV<br />
When a monatomic ideal gas is heated at constant volume, all of the heat goes into increasing<br />
the translational kinetic energy of the atoms. However, in the case of molecules, there are other<br />
energy modes available in addition to kinetic energy. Specifically, molecules may have rotational<br />
and vibrational energy. Therefore, when a molecular gas is heated, the resulting energy increase is<br />
distributed among all three energy modes. Statistically, it may be shown that the amount of energy<br />
per mole that goes into each energy mode is<br />
U m = N m(½RT),<br />
where N is related to the number of degrees of freedom and the number of different types of energy<br />
m<br />
(e.g., kinetic and potential) associated with the energy mode, and R is the gas constant. This result,<br />
which assumes that all energy modes have continuous energy distributions, is known as the<br />
equipartition theorem. The number of degrees of freedom for a molecule, D , is related to the<br />
m<br />
number of coordinates required to specify the position of each atom. The degrees of freedom<br />
associated with the various energy modes of a molecule and the corresponding values of N are m<br />
summarized below;<br />
N = D , N = D , N = 2D<br />
tran tran rot rot vib vib<br />
D = 3n tot<br />
D = 3 tran<br />
D = 2 (linear molecule) or 3 (nonlinear)<br />
rot<br />
D = 3n !5 (linear molecule) or 3n ! 6 (nonlinear)<br />
vib<br />
The equipartition theorem may be used to estimate the heat capacities of gas-phase molecules at high<br />
temperatures.<br />
Example: Estimate the value of C for CO .<br />
V 2
4-11<br />
In this case, n = 3 and so D = 9. This molecule is linear; therefore D = 2 and D<br />
tot rot vib<br />
= 9 ! 5 = <strong>4.</strong> Hence U = U tran + U rot + U vib = 3(½RT) + 2(½RT) + 8(½RT) =<br />
13/2 RT. The estimated heat capacity is therefore C V<br />
' dU<br />
dT ' 13 R ' 6.5 R. The<br />
2<br />
measured value at 298 K is 3.46 R.<br />
Exercise: Estimate the value of C for CH .<br />
V 4<br />
In this case, n = 5 and so D tot = 15. This molecule is nonlinear; therefore D rot = 3 and<br />
D vib = 15 ! 6 = 9. Hence U = U tran + U rot + U vib = 3(½RT) + 3(½RT) + 18(½RT) =<br />
12RT. The estimated heat capacity is therefore C V<br />
' dU ' 12 R. The measured<br />
dT<br />
value at 298 K is 3.25 R.<br />
Because the energies of rotation and vibration are not really continuous, the values of C obtained<br />
V<br />
for molecules from the equipartition theorem are reached only at very high temperatures.<br />
The relationship between C V and C P can be derived for an ideal gas as follows:<br />
H = U + PV<br />
MH<br />
' MU % P MV<br />
MT P<br />
MT P<br />
MT<br />
P<br />
,<br />
but for an ideal gas, the internal energy only depends on temperature U = f(T) (recall that U = trans<br />
(3/2)RT), and so<br />
MU<br />
' MU<br />
MT P<br />
MT<br />
' C V<br />
.<br />
V<br />
Also<br />
V m<br />
' RT<br />
P , so MV m<br />
MT<br />
' R . Therefore<br />
P<br />
P<br />
.<br />
Exercise:<br />
Determine the values of C V and C P for Ar assuming it to be an ideal gas.<br />
U = (3/2)RT so C V = dU/dT = (3/2)R and C P = (5/2)R.
4-12<br />
Exercise:<br />
Draw a P-V diagram for one mole of a monatomic ideal gas initially at a pressure of<br />
2.5 atm and a volume of 5.0 L undergoing the following: (a) the pressure is reduced<br />
suddenly to 1.0 atm while the volume is held constant, (b) then, the volume expands<br />
to 12.5 L while the pressure is held constant. Calculate U, H, q, and w for both<br />
steps and for the isothermal, reversible expansion from the initial to the final state.<br />
T = P V /R = 5(2.5)/0.08206 =152.3K<br />
1 1 1<br />
T = 5(1.0)/0.08206 = 60.9 K<br />
2<br />
T = T = 152.3 K<br />
3 1<br />
P (atm)<br />
3.0<br />
2.5<br />
2.0<br />
1.5<br />
1.0<br />
0.5<br />
(5.0, 2.5)<br />
A<br />
C<br />
B<br />
(5.0, 1.0) (12.5, 1.0)<br />
Step a:<br />
U = C T = (3/2)(8.314)(!91.4)<br />
V<br />
= !1139.8 J<br />
0.0<br />
4 6 8 10 12 14<br />
V (L)<br />
H = U + (PV) = !1139.8 +[(1.0)(5.0)!(2.5)(5.0)] x 8.314/0.08206<br />
= !1899.7 J Also H = C T = 5/2(8.314)(!91.4) = !1899.7 J (state func.)<br />
P<br />
w = 0, q = U<br />
Step b:<br />
H = C T = (5/2)(8.314)(91.9) = 1899.7 J<br />
Step c:<br />
q = H<br />
P<br />
w = !P V = !(1.0)(12.5 ! 5.0) x 8.314/0.08206 = !759.9 J<br />
U = q + w = 1899.7 ! 759.9 = 1139.8 J Also U = C T = 3/2(8.314)(91.4)<br />
V<br />
U = 0<br />
(T is constant)<br />
H = U + (PV) = 0 (P V = P V )<br />
1 1 2 2<br />
w = !RT ln V /V = !(8,314)(152.3) ln (12.5/5.0) = !1160.2 J<br />
2 1<br />
q = !w<br />
= 1139.8 J (state function)<br />
(Note that both U and H are zero for the closed cycle 16 2 6 1).
F. Thermochemistry<br />
4-13<br />
A very important application of thermodynamics to chemistry is the study of the heat released<br />
or absorbed in a chemical reaction. Chemical reactions are most commonly carried out at constant<br />
pressure (i.e., in an open vessel) and for this specified path, the heat changes are the same as the<br />
enthalpy changes. Since enthalpy is a state function, it is possible to calculate the change in enthalpy<br />
accompanying a chemical reaction simply from a knowledge of the enthalpies of the reactants and<br />
the enthalpies of the products. In actual, practice, enthalpies for chemical reactions may be<br />
o<br />
determined from tabulated standard enthalpy changes, denoted by the symbol H . The term<br />
standard means that the enthalpy change is for a reaction in which the reactants and products are in<br />
their so-called standard states.<br />
Standard state:<br />
the pure substance at a specified<br />
temperature and a pressure of 1 bar.<br />
Although enthalpy changes may be tabulated for any temperature, the most common tabulations are<br />
for 298 K.<br />
1. Enthalpy of formation<br />
The most useful thermochemical tabulations give standard enthalpies of formation denoted<br />
o<br />
by the symbol fH .<br />
Standard enthalpy of formation:<br />
the enthalpy change accompanying the formation of a<br />
substance in its standard state from the most stable forms of<br />
the constituent elements of the substance in their standard<br />
states.<br />
For example, the standard enthalpy of formation for benzene is the enthalpy change for the reaction<br />
6C + 3H ! C H .<br />
(s,gr) 2(g) 6 6(l)
is<br />
=<br />
=<br />
=<br />
4-14<br />
At 298 K and 1 bar, where the solid (graphite) is the most stable form of carbon, the diatomic gas<br />
is the most stable form of hydrogen, and the liquid is the most stable form of benzene, the standard<br />
o<br />
o<br />
enthalpy of formation of benzene is H (C H ) = 49.0 kJ/mol. Note that H is a molar quantity<br />
f 6 6 f<br />
and the formation reaction is always balanced such that one mole of product is formed. The<br />
enthalpies of formation of all pure elements in their standard states are taken to be zero.<br />
of formation:<br />
o<br />
Now consider the task of calculating H for the following reaction using standard enthalpies<br />
(1) 2HN + 2NO ! H O + 4N .<br />
3(l) (g) 2 2(l) 2(g)<br />
We begin by writing the formation reactions for each of the (nonelemental) reactants and products<br />
and looking up the standard enthalpies of formation;<br />
o<br />
(2) 3/2 N 2(g) + 1/2 H 2(g) ! HN 3(l) fH (HN 3) = 26<strong>4.</strong>0 kJ/mol<br />
o<br />
(3) 1/2 N 2(g) + 1/2 O 2(g) ! NO (g) fH (NO) = 90.25 kJ/mol<br />
o<br />
(4) H 2(g) + O 2(g) ! H2O 2(l) fH (H2O 2) = !187.78 kJ/mol<br />
Next, we rearrange the reactions in such a way that they add up to reaction (1);<br />
Therefore,<br />
reaction (1) = !2 x reaction (2) ! 2 x reaction (3) + reaction (4) .<br />
o o o o<br />
H = fH (H2O 2) ! 2 fH (HN 3) !2 fH (NO) = !896.28 kJ/mol.<br />
This is an illustration of Hess's Law. The above procedure may be generalized and stated in a more<br />
concise form by representing each of the stoichiometric coefficients of the net reaction by the symbol<br />
ν, where i represents the reaction species and the sign of taken to be positive for products and<br />
ν<br />
i<br />
negative for reactants. (For example, in reaction 1 above,<br />
ν<br />
i<br />
1,<br />
ν<br />
!2, and<br />
ν<br />
1 2 3<br />
!2). Then<br />
.
4-15<br />
Applying this formula directly to reaction (1) above gives<br />
o o o o<br />
H = fH (H2O 2) ! 2 fH (HN 3) !2 fH (NO) = !896.28 kJ/mol,<br />
which is exactly the same as we obtained before.<br />
Hess's law is a direct consequence of the fact that enthalpy is a state function; the enthalpy<br />
change for a reaction is the same for any path that connects the reactants and products.<br />
Exercise:<br />
Construct a diagram for the above example showing the relationship between the<br />
direct reaction path and the path involving the formation reactions.<br />
2HN + 2NO !<br />
H O + 4N<br />
3 2 2 2<br />
9 9 8<br />
(3N + H ) + (N + O ) ! (4N + H + O ) .<br />
2 2 2 2 2 2 2<br />
Exercise:<br />
Use the tabulated enthalpies of formation in your textbook to calculate the enthalpy<br />
of combustion for phenol (C H OH).<br />
6 5<br />
C H OH + 7O ! 6CO + 3H O<br />
6 5 (s) 2(g) 2(g) 2 (l)<br />
o o o o<br />
cH (C6H5OH) = 6 fH (CO 2) + 3 fH (H2O) ! fH (C6H5OH)<br />
2. Enthalpies of physical change<br />
= 6(!393.51) + 3(!285.83) ! (!165.0) = !3054 kJ/mol.<br />
Other enthalpy changes of importance in chemistry are those that accompany transitions from<br />
one physical form to another. The transitions of particular interest are defined below.<br />
Transition Physical change Symbol for enthalpy change<br />
Fusion solid to liquid H fus<br />
Vaporization liquid to gas H vap<br />
Sublimation solid to gas H sub<br />
Hydration gaseous ions to ions H hyd<br />
in solution<br />
Solution solute to solution H sol
4-16<br />
Ionization gaseous atom to H ion<br />
gaseous positive ion<br />
Electron gain gaseous atom to H eg<br />
Gaseous negative ion<br />
Bond dissociation gaseous molecule to H dis<br />
dissociated gaseous species<br />
G. Bond Enthalpies<br />
One consequence of the fact that bond formation in many molecules is highly localized<br />
between neighboring atoms is that bond enthalpies are fairly insensitive to the other (non-neighbor)<br />
atoms of the molecule. As a result of this, average bond enthalpies that have been determined for<br />
bonds between various atoms may be used to estimate enthalpies of reactions. Average bond<br />
enthalpies for bonds of H, C, N, and O atoms with other common elements are given in Table 4-1.<br />
Table 4-1. Bond enthalpies of common elements (kJ/mol)<br />
H! C! C= C/ N! N= N/ O! O=<br />
H 436 413 391 463<br />
C 413 348 615 812 292 615 891 351 728<br />
N 391 292 615 891 161 418 946<br />
O 463 351 728 139 485<br />
F 563 441 270 185<br />
Si 295 290 369<br />
P 320<br />
S 339 259 477<br />
Cl 432 328 200 203<br />
Br 366 276<br />
I 299 240<br />
Bond enthalpies, such as those given in the above table, may be determined from enthalpies<br />
of formation. For example, to obtain the C!H bond enthalpy, One can use the following information<br />
for the CH molecule: CH ! C + 4H<br />
4 4(g) (g) (g)<br />
o<br />
H (CH<br />
f<br />
4(g)<br />
) = !7<strong>4.</strong>85 kJ/mol
orbitals<br />
orbitals<br />
4-17<br />
o<br />
H (C ) = 718.38 kJ/mol<br />
f<br />
(g)<br />
o<br />
H (H ) = 217.94 kJ/mol.<br />
f<br />
o<br />
Then H = 718.38 + 4(217.94)+ 7<strong>4.</strong>85 = 1665.0 kJ,<br />
and therefore the average C!H bond energy is 1665.0/4 = 416.0 kJ/mol.<br />
(g)<br />
As an illustration of how bond enthalpies may be used to estimate enthalpies of formation,<br />
consider the case of benzene;<br />
6C + 3H ! C H .<br />
(gr) 2(g) 6 6(g)<br />
Using the tabulated C=C, C!C, and C!H bond enthalpies, the<br />
formation of benzene from the gaseous atoms as follows:<br />
o<br />
H can be estimated for the<br />
Form 6 C!H bonds = 6 x !413 = !2478 kJ (Note: When bonds<br />
3 C!C bonds = 3 x !348 = !1044 kJ are formed, H is<br />
3 C=C bonds = 3 x !615 = !1845 kJ negative).<br />
Total bond formation = !5367 kJ.<br />
Then, to obtain the enthalpy of formation, we must add the enthalpy changes for the additional two<br />
steps;<br />
o<br />
o<br />
6C 6 6C , H = 6 H (C ) = 6 x 718.38 = 4308 kJ/mol<br />
(gr) (g) f (g)<br />
o<br />
o<br />
3H 6 6H , H = 6 H (H ) = 6 x 217.94 = 1308 kJ/mol.<br />
2(g) (g) f (g)<br />
o<br />
Therefore, fH (C6H 6(g) ) = !5367 + 4308 + 1308 = 249 kJ/mol. Actually, this turns out to be a rather<br />
poor estimate (the experimental value is 83 kJ/mol) because the double bonds in benzene are best<br />
2<br />
described in terms of sp hybridized σ<br />
and delocalized<br />
π<br />
that extend around the<br />
whole molecule. The delocalization causes the bond enthalpy to be considerably larger than that<br />
expected for single and double bonds. This reduction in the bond enthalpy is referred to as<br />
resonance stabilization.<br />
H. Temperature dependence of enthalpy<br />
It is often necessary to know reaction enthalpies at temperatures other than 298 K.<br />
Consider the cycle shown below involving the same reaction at two temperatures T and T .<br />
1 2
4-18<br />
Since H is a state function<br />
H(T ) 2<br />
aA + bB ! cC T 2<br />
H 9 H 9 8 H<br />
1 2 3<br />
aA + bB ! cC T 1<br />
H(T ) 1<br />
H(T ) = H + H + H(T ) + H .<br />
2 1 2 1 3<br />
For a reaction at constant pressure, the<br />
substance may be calculated from the relation<br />
H associated with changing the temperature of each<br />
T 2<br />
H ' n C P<br />
dT.<br />
T 1<br />
Therefore, T 1<br />
T 1<br />
T 2<br />
H(T 2) = H(T 1) + a C P<br />
(A)dT % b C P<br />
(B)dT % c C P<br />
(C)dT.<br />
T 2<br />
T 2<br />
T 1<br />
Over a restricted range of temperatures, the heat capacities may be treated as constants, in which case<br />
the above formula reduces to<br />
where<br />
H(T ) = H(T ) + C (T ! T ),<br />
2 1 P 2 1<br />
C j C P (i)<br />
P = cC P(C) ! aC P(A) ! bC P(B) = .<br />
iνi<br />
I. Work of adiabatic expansion<br />
The work associated with the expansion of a gas is given by<br />
V 2<br />
w ' !<br />
V 1<br />
P ex<br />
dV<br />
,<br />
and for the isothermal expansion of an ideal gas, we found<br />
a). w = !P V<br />
ex<br />
when P = constant<br />
ex
). w = !nRT ln (V /V )<br />
2 1<br />
4-19<br />
when process is reversible.<br />
Also, for an ideal gas, since U = 0 when T = 0,<br />
q = !w.<br />
Now we shall explore the properties of an adiabatic expansion. For an adiabatic expansion,<br />
q = 0, and so dU = dw. Therefore,<br />
w ' dU ,<br />
which means that when a gas expands adiabatically, the work done on the surroundings is<br />
accompanied by a decrease in the internal energy of the system. Hence, the temperature of an ideal<br />
gas will decrease in an adiabatic expansion. Since U is a state function, we may obtain the work for<br />
an adiabatic expansion of n moles of an ideal gas from<br />
T 2<br />
w ' U ' n C V<br />
dT ' nC V<br />
T,<br />
T 1<br />
where the last expression assumes C is constant over the temperature range. The above equation<br />
V<br />
applies to both reversible and irreversible processes.<br />
Lets consider the important case of a reversible adiabatic expansion of an ideal gas. The<br />
initial state of the system is specified by P , V , and T , while the final state of the system is specified<br />
1 1 1<br />
by P , V , and T .<br />
2 2 2<br />
so<br />
Therefore,<br />
and<br />
a). Determine the relationship between V and T.<br />
where c = C /R. V<br />
Thus<br />
dw = !PexdV = !(nRT/V)dV = nCVdT,<br />
ln V 1<br />
V 2<br />
T 2<br />
C V<br />
dT<br />
T<br />
T 1<br />
V 2<br />
RdV<br />
' ! .<br />
V<br />
V 1<br />
C V<br />
ln T 2<br />
T 1<br />
' Rln V 1<br />
V 2<br />
' C V<br />
R ln T 2<br />
T 1<br />
' ln T 2<br />
T 1<br />
,<br />
c<br />
,
=<br />
><br />
=<br />
4-20<br />
.<br />
b). Determine the relationship between P and V.<br />
P 1<br />
V 1<br />
T 1<br />
' P 2 V 2<br />
T 2<br />
so<br />
Now<br />
P 1<br />
V 1<br />
' T 1<br />
' V 1<br />
2 c<br />
P 2<br />
V 2<br />
T 2<br />
V 1<br />
1% 1 c<br />
P 1<br />
V1 ' P 2<br />
V<br />
1% 1 c<br />
1 % 1 c ' 1 % R C V<br />
' C V % R<br />
2<br />
.<br />
C V<br />
Let γ<br />
C P/C V, then<br />
' C P<br />
C V<br />
.<br />
.<br />
Note that γ<br />
1 for all gases and γ<br />
5/3 for a monatomic ideal gas.<br />
' T 1<br />
Exercise: Show that ,<br />
P 2<br />
where<br />
P 1<br />
T 2<br />
c )<br />
c’ = C /R. P<br />
P 1<br />
V 1<br />
P 2<br />
V 2<br />
' T 1<br />
T 2<br />
P 1<br />
P 2<br />
T 2<br />
T 1<br />
c<br />
' T 1<br />
T 2<br />
P 1<br />
P 2<br />
' T 1<br />
T 2<br />
1%c<br />
' T 1<br />
T 2<br />
c )<br />
.
4-21<br />
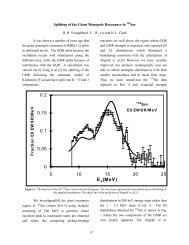
An adiabatic expansion is shown schematically on the PVT surface in Fig. 4-4, and an<br />
adiabat (graph of P versus V for an adiabatic process) is compared with an isotherm (graph of P<br />
versus V for an isothermal process) in Fig. 4-5. It should be noted in Fig. 4-4 that the pressure<br />
decreases more in an adiabatic expansion than in an isothermal expansion between the same initial<br />
and final volumes.<br />
Figure <strong>4.</strong><strong>4.</strong> Depiction of an isothermal and an adiabatic expansion on the PVT surface (From<br />
th<br />
Physical Chemistry, 5 Edition, Peter Atkins, W. H. Freeman and Co., New York,<br />
1994).
4-22<br />
1.2<br />
1.1<br />
1.0<br />
0.9<br />
P (atm)<br />
0.8<br />
0.7<br />
0.6<br />
Isotherm<br />
0.5<br />
0.4<br />
0.3<br />
Adiabat<br />
0.2<br />
20 25 30 35 40 45 50<br />
V (L)<br />
Figure 4-5. An isotherm and an adiabat for the expansion of one mole of a monatomic ideal gas.
4-23<br />
Review Questions<br />
1. Define open, closed, and isolated thermodynamic systems.<br />
2. What is a state function? List some state functions. List some non-state functions.<br />
3. Define a reversible process.<br />
<strong>4.</strong> Calculate the work involved in each of the following:<br />
a).<br />
b).<br />
A balloon containing 1 mole of O at 298 K is suddenly ejected into outer space from the<br />
2<br />
space shuttle. (Neglect the elastic force of the balloon).<br />
A bubble in a bottle of champagne expands when the bottle is uncorked. Assume the<br />
bubble contains 0.3 mg of CO at 1.5 atm and 5 C, and that the outside pressure is 1 atm).<br />
2<br />
c). The expansion in part b is carried out reversibly.<br />
5. Draw a PV diagram for parts (b) and (c) of problem <strong>4.</strong><br />
6. A sufficient amount of heat is transferred to 25.0 g of carbon tetrachloride at its boiling<br />
temperature (76.1 C) to cause it to evaporate inside a container with a frictionless piston. The<br />
external pressure is 1 atm.<br />
a).<br />
b).<br />
c).<br />
d).<br />
Does all the heat get changed into internal energy?<br />
If your answer to (a) was no, explain where the rest goes.<br />
The heat of vaporization of CCl is 30.00 kJ/mol. Calculate the internal energy change for<br />
4<br />
this process. (Neglect the volume of the liquid).<br />
After the CCl has evaporated, the outside pressure is reduced to 0.5 atm while the<br />
4<br />
temperature is kept constant. Calculate the change in the internal energy and the change in<br />
enthalpy associated with the resulting expansion.<br />
7. Derive the relationship between C P and C V for an ideal gas.<br />
8. Estimate the high temperature value of C for water vapor.<br />
V<br />
9. How much heat must be transferred to 3 mole of Ar to raise its temperature from 25 C to 100 C<br />
(a) at constant volume, and (b) at constant pressure?<br />
10. The standard heat of combustion of 1-hexene is -4034 kJ/mol. Calculate its standard heat of<br />
formation.<br />
11. Give a physical explanation of why C P is larger than C V.
4-24<br />
12. Write reactions to define each of the following enthalpies:<br />
(a).<br />
sublimation<br />
(b). solution<br />
(c).<br />
hydration<br />
(d). ionization<br />
(e).<br />
lattice<br />
13. Use tabulated bond enthalpies to estimate the enthalpy for the reaction<br />
C H + H ! C H .<br />
2 4(g) 2(g) 2 6(g)<br />
1<strong>4.</strong> Calculate H for the freezing of 2.000 mol of supercooled liquid water at -15.00 C at a pressure<br />
of 1.00 atm, given that the standard enthalpy of fusion (T = 0 C) is 6.008 kJ/mol. Assume the<br />
heat capacities of liquid water (76.1 J/mol-K) and ice (37.15 J/mol-K) are constant.<br />
15. A 28.0 g sample of Kr at 25 atm and 300 K expands reversibly and adiabatically to a final<br />
volume of 10.0 L. Calculate the following assuming Kr is an ideal gas:<br />
(a).<br />
(b). U<br />
(c).<br />
The final temperature<br />
q<br />
(d). w<br />
(e). H .<br />
16. A 28.0 g sample of Kr at 25 atm and 300 K expands reversibly and adiabatically to a final<br />
pressure of 2.00 atm. Calculate the following assuming Kr is an ideal gas:<br />
(a).<br />
(b). U<br />
(c).<br />
The final temperature<br />
q<br />
(d). w<br />
(e). H .
Answers:<br />
4-25<br />
<strong>4.</strong> (a) w = 0, (b) w = -0.0053 J, (c) w = -0.0064 J<br />
rev<br />
6. (c) U = <strong>4.</strong>40 kJ, (d) H = 0.24 kJ<br />
8. 6R<br />
9. (a) q =2.806 kJ, (b) q = <strong>4.</strong>677 kJ<br />
o<br />
10. fH (C6H 12) = -42 kJ/mol<br />
o<br />
13. H = !123 kJ/mol<br />
o<br />
1<strong>4.</strong> fusH (258) = !10.848 kJ<br />
15. a) T = 30.8 K, (b) U = -1121 J, (c) q = 0, (d) w = -1121 J, (e) H = -1869 J<br />
2<br />
16. a) T = 109 K , (b) U = -79<strong>4.</strong>6 J, (c) q = 0, (d) w = -79<strong>4.</strong>6 J, (e) H = -1325 J<br />
2
4-26<br />
FORMULAS YOU SHOULD KNOW<br />
First law of thermodynamics:<br />
U ' q % w<br />
Differential work of expansion:<br />
Work for isothermal reversible<br />
expansion of an ideal gas:<br />
Enthalpy:<br />
Heat capacity<br />
w ' !<br />
C V<br />
' dq V<br />
dT '<br />
C P<br />
' dq P<br />
dT '<br />
dw ' !P ex<br />
dV<br />
V 2<br />
V 1<br />
P int<br />
dV ' !nRT<br />
H ' U % PV<br />
constant volume: ;<br />
Constant pressure: ;<br />
V 2<br />
MU U ' n<br />
MT V<br />
MH H ' n<br />
MT P<br />
dV<br />
V ' !nRT ln V 2<br />
V<br />
V 1<br />
1<br />
T 2<br />
T 1<br />
C V<br />
dT . nC V<br />
T<br />
T 2<br />
T 1<br />
C P<br />
dT . nC P<br />
T<br />
Relationship between C V and CP<br />
for an ideal gas:<br />
C P<br />
' C V<br />
% R<br />
Hess’s law:<br />
H o ' jiνi f H o (i)<br />
Reversible adiabatic expansion of an ideal gas:<br />
w ' n<br />
V 1<br />
V 2<br />
' T 2<br />
T 1<br />
T 2<br />
T 1<br />
C V<br />
dT ' nC V<br />
T<br />
c<br />
or<br />
T 2<br />
' V 1<br />
1 c<br />
T 1<br />
V 2
=<br />
4-27<br />
where<br />
where<br />
c = C /R. V<br />
P 1<br />
P 2<br />
' T 1<br />
T 2<br />
c )<br />
c’ = C /R<br />
,<br />
P<br />
,<br />
P 1<br />
Vγ1 ' P 2Vγ2 ' constant<br />
where γ<br />
C P/C V.