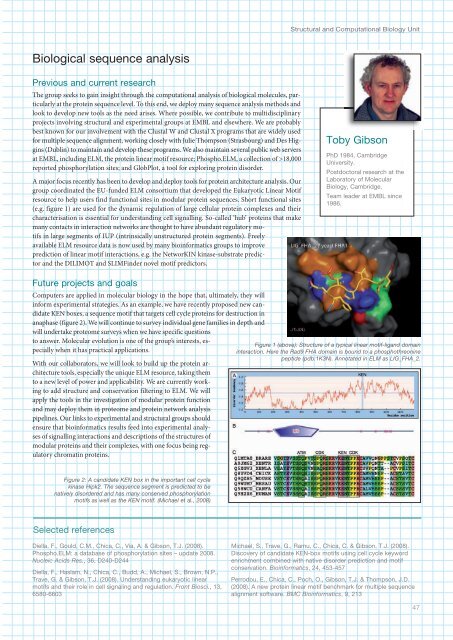
Structural and Computational Biology UnitBiological sequence analysisPrevious and current researchThe group seeks to gain insight through the computational analysis of biological molecules, particularlyat the protein sequence level. To this end, we deploy many sequence analysis methods andlook to develop new tools as the need arises. Where possible, we contribute to multidisciplinaryprojects involving structural and experimental groups at <strong>EMBL</strong> and elsewhere. We are probablybest known for our involvement with the Clustal W and Clustal X programs that are widely usedfor multiple sequence alignment, working closely with Julie Thompson (Strasbourg) and Des Higgins(Dublin) to maintain and develop these programs. We also maintain several public web serversat <strong>EMBL</strong>, including ELM, the protein linear motif resource; Phospho.ELM, a collection of >18,000reported phosphorylation sites; and GlobPlot, a tool for exploring protein disorder.A major focus recently has been to develop and deploy tools for protein architecture analysis. Ourgroup coordinated the EU-funded ELM consortium that developed the Eukaryotic Linear Motifresource to help users find functional sites in modular protein sequences. Short functional sites(e.g. figure 1) are used for the dynamic regulation of large cellular protein complexes and theircharacterisation is essential for understanding cell signalling. So-called ‘hub’ proteins that makemany contacts in interaction networks are thought to have abundant regulatory motifsin large segments of IUP (intrinsically unstructured protein segments). Freelyavailable ELM resource data is now used by many bioinformatics groups to improveprediction of linear motif interactions, e.g. the NetworKIN kinase-substrate predictorand the DILIMOT and SLIMFinder novel motif predictors.Toby GibsonPhD 198, CambridgeUniversity.Postdoctoral research at theLaboratory of MolecularBiology, Cambridge.Team leader at <strong>EMBL</strong> since1986.Future projects and goalsComputers are applied in molecular biology in the hope that, ultimately, they willinform experimental strategies. As an example, we have recently proposed new candidateKEN boxes, a sequence motif that targets cell cycle proteins for destruction inanaphase (figure 2). We will continue to survey individual gene families in depth andwill undertake proteome surveys when we have specific questionsto answer. Molecular evolution is one of the group’s interests, especiallywhen it has practical applications.With our collaborators, we will look to build up the protein architecturetools, especially the unique ELM resource, taking themto a new level of power and applicability. We are currently workingto add structure and conservation filtering to ELM. We willapply the tools in the investigation of modular protein functionand may deploy them in proteome and protein network analysispipelines. Our links to experimental and structural groups shouldensure that bioinformatics results feed into experimental analysesof signalling interactions and descriptions of the structures ofmodular proteins and their complexes, with one focus being regulatorychromatin proteins.Figure 1 (above): Structure of a typical linear motif-ligand domaininteraction. Here the Rad9 FHA domain is bound to a phosphothreoninepeptide (pdb:1K3N). Annotated in ELM as LIG_FHA_2.Figure 2: A candidate KEN box in the important cell cyclekinase Hipk2. The sequence segment is predicted to benatively disordered and has many conserved phosphorylationmotifs as well as the KEN motif. (Michael et al., 2008)Selected referencesDiella, F., Gould, C.M., Chica, C., Via, A. & Gibson, T.J. (2008).Phospho.ELM: a database of phosphorylation sites – update 2008.Nucleic Acids Res., 36, D20-D2Diella, F., Haslam, N., Chica, C., Budd, A., Michael, S., Brown, N.P.,Trave, G. & Gibson, T.J. (2008). Understanding eukaryotic linearmotifs and their role in cell signaling and regulation. Front Biosci., 13,6580-6603Michael, S., Trave, G., Ramu, C., Chica, C. & Gibson, T.J. (2008).Discovery of candidate KEN-box motifs using cell cycle keywordenrichment combined with native disorder prediction and motifconservation. Bioinformatics, 2, 53-57Perrodou, E., Chica, C., Poch, O., Gibson, T.J. & Thompson, J.D.(2008). A new protein linear motif benchmark for multiple sequencealignment software. BMC Bioinformatics, 9, 2137
<strong>EMBL</strong> Research at a Glance 2009Edward LemkePhD, MPI for BiophysicalChemistry, Göttingen.Research Associate, theScripps Research Institute.Group leader at <strong>EMBL</strong> since2009. Joint appointment withCell Biology and BiophysicsUnit.Structural light microscopy/single moleculespectroscopyPrevious and current researchResearch in our laboratory combines modern chemical biology and biochemistry/molecular biologymethods with advanced fluorescence and single molecule techniques to elucidate the natureof protein disorder in biological systems and disease mechanisms.Currently, more than 50,000 protein structures with atomic resolution are available from the proteindatabank and due to large efforts (mainly crystallography and NMR) their number is rapidlygrowing. However, even if all 3D protein structures were available, our view of the molecular buildingblocks of cellular function will still be rather incomplete, as we now know that many proteinsare intrinsically disordered, which means that they are unfolded in their native state. Interestingly,the estimated percentage of intrinsically disordered proteins (IDPs) grows with the complexity ofthe organism (prokaryotes ≈ 5% and eukaryotes ≈ 50%). In a modern view of systems biology, thesedisordered proteins are believed to be multi-functional signalling hubs central to the interactome(the whole set of molecular interactions in the cell). Their ability to adopt multiple conformationsis considered a major driving force behind their evolution and enrichment in eukaryotes.While the importance of IDPs in biology is now well established, many common strategies for probing protein structure are incompatible withmolecular disorder and the highly dynamic nature of those systems. In addition, a mosaic of molecular states and reaction pathways can existin parallel in any complex biological system, further complicating the situation to measure these systems. For example, some proteins mightbehave differently than the average, giving rise to new and unexpected phenotypes. One such example are the infamous Prion proteins, wheremisfolding of only subpopulations of proteins can trigger a drastic signalling cascade leading to completely new phenotypes. Conventionalensemble experiments are only able to measure the average behaviour of a system, discounting such coexisting populations and rare events.Ignoring such information can easily lead to generation of false or insufficient models, which may further impede our understanding of thebiological processes and disease mechanisms.In contrast, single molecule techniques, which probe the distribution of behaviours, can shed light on important mechanisms that otherwise remainmasked. In particular, single molecule fluorescence (smF) studies allow probing of molecular structures and dynamics on the nanometerscale with high time resolution. Although not inherently limited bythe size of a macromolecule, smF studies require site-specific labellingwith special fluorescent dyes which still hampers the broad applicationand general use of this technique. It was recently demonstratedthat amber nonsense suppression technology of genetically reprogrammedhosts is an especially powerful approach to overcome thislimitation (Brustad et al., 2008). Here, unnatural amino acids withunique chemical properties are conveniently site-specifically introducedinto any protein site by the host organism itself, serving as manipulationsites. Our lab also continues to develop and apply suchprotein engineering tools to facilitate fluorescence studies of complexbiological mechanisms.Labelled proteins are excited using advanced laser techniques andemitted fluorescence photons are detected using home-built highlysensitive equipment. This strategy allows to study structure andFuture projects and goalsdynamics of even heterogeneous biological systems.Recent studies have shown that even the building blocks of some ofthe most complex and precise machines with an absolute critical role to survival of the cell, such as DNA packing and many transport processes,are largely built from IDPs. We aim to explore the physical and molecular rationale behind the fundamental role of IDPs by combining molecularbiology and protein engineering tools with single molecule biophysics. Our long-term goal is to develop general strategies to study structureand dynamics of IDPs within their natural complex environments.Selected referencesFerreon, A.C.M., Gambin, Y, Lemke, E.A., Deniz A.A. (2009). Single-Molecule Fluorescence illuminates a multi-conformational switch inα-synuclein. Proc. Natl. Acad. Sci. USA, doi:10.1073/pnas.0809232106Brustad, E.M., Lemke, E.A., Schultz, P.G. & Deniz, A.A. (2008). Ageneral and efficient method for the site-specific dual-labeling ofproteins for single molecule fluorescence resonance energy transfer.J. Am. Chem. Soc., 130, 1766-58Deniz, A.A., Mukhopadhyay, S. & Lemke, E.A. (2008). Singlemoleculebiophysics: at the interface of biology, physics andchemistry. J. R. Soc. Interface, 5, 15-5Lemke, E.A., Summerer, D., Geierstanger, B.H., Brittain, S.M. &Schultz, P.G. (2007). Control of protein phosphorylation with agenetically encoded photocaged amino acid. Nat. Chem. Biol., 3,769-72