

Oxygen isotope biogeochemistry of pore water sulfate in the deep ...
Oxygen isotope biogeochemistry of pore water sulfate in the deep ...
Oxygen isotope biogeochemistry of pore water sulfate in the deep ...
- No tags were found...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
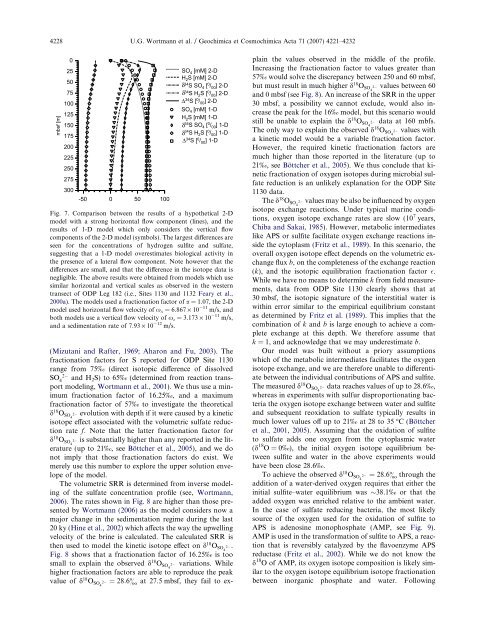
4228 U.G. Wortmann et al. / Geochimica et Cosmochimica Acta 71 (2007) 4221–4232mbsf [m]0255075100125150175200225250275300-50 0 50 100SO 4 [mM] 2-DH 2 S [mM] 2-Dδ 34 S SO 4 [ 0 / 00 ] 2-Dδ 34 S H 2 S [ 0 / 00 ] 2-DΔ 34 S [ 0 / 00 ] 2-DSO 4 [mM] 1-DH 2 S [mM] 1-Dδ 34 S SO 4 [ 0 / 00 ] 1-Dδ 34 S H 2 S [ 0 / 00 ] 1-DΔ 34 S [ 0 / 00 ] 1-DFig. 7. Comparison between <strong>the</strong> results <strong>of</strong> a hypo<strong>the</strong>tical 2-Dmodel with a strong horizontal flow component (l<strong>in</strong>es), and <strong>the</strong>results <strong>of</strong> 1-D model which only considers <strong>the</strong> vertical flowcomponents <strong>of</strong> <strong>the</strong> 2-D model (symbols). The largest differences areseen for <strong>the</strong> concentrations <strong>of</strong> hydrogen sulfite and <strong>sulfate</strong>,suggest<strong>in</strong>g that a 1-D model overestimates biological activity <strong>in</strong><strong>the</strong> presence <strong>of</strong> a lateral flow component. Note however that <strong>the</strong>differences are small, and that <strong>the</strong> difference <strong>in</strong> <strong>the</strong> <strong>isotope</strong> data isnegligible. The above results were obta<strong>in</strong>ed from models which usesimilar horizontal and vertical scales as observed <strong>in</strong> <strong>the</strong> westerntransect <strong>of</strong> ODP Leg 182 (i.e., Sites 1130 and 1132 Feary et al.,2000a). The models used a fractionation factor <strong>of</strong> a = 1.07, <strong>the</strong> 2-Dmodel used horizontal flow velocity <strong>of</strong> x x = 6.867 · 10 11 m/s, andboth models use a vertical flow velocity <strong>of</strong> x z = 3.173 · 10 11 m/s,and a sedimentation rate <strong>of</strong> 7.93 · 10 12 m/s.(Mizutani and Rafter, 1969; Aharon and Fu, 2003). Thefractionation factors for S reported for ODP Site 1130range from 75‰ (direct isotopic difference <strong>of</strong> dissolved2SO 4and H 2 S) to 65‰ (determ<strong>in</strong>ed from reaction transportmodel<strong>in</strong>g, Wortmann et al., 2001). We thus use a m<strong>in</strong>imumfractionation factor <strong>of</strong> 16.25‰, and a maximumfractionation factor <strong>of</strong> 57‰ to <strong>in</strong>vestigate <strong>the</strong> <strong>the</strong>oreticald 18 O SO4 2 evolution with depth if it were caused by a k<strong>in</strong>etic<strong>isotope</strong> effect associated with <strong>the</strong> volumetric <strong>sulfate</strong> reductionrate f. Note that <strong>the</strong> latter fractionation factor ford 18 O SO4 2 is substantially higher than any reported <strong>in</strong> <strong>the</strong> literature(up to 21‰, see Böttcher et al., 2005), and we donot imply that those fractionation factors do exist. Wemerely use this number to explore <strong>the</strong> upper solution envelope<strong>of</strong> <strong>the</strong> model.The volumetric SRR is determ<strong>in</strong>ed from <strong>in</strong>verse model<strong>in</strong>g<strong>of</strong> <strong>the</strong> <strong>sulfate</strong> concentration pr<strong>of</strong>ile (see, Wortmann,2006). The rates shown <strong>in</strong> Fig. 8 are higher than those presentedby Wortmann (2006) as <strong>the</strong> model considers now amajor change <strong>in</strong> <strong>the</strong> sedimentation regime dur<strong>in</strong>g <strong>the</strong> last20 ky (H<strong>in</strong>e et al., 2002) which affects <strong>the</strong> way <strong>the</strong> upwell<strong>in</strong>gvelocity <strong>of</strong> <strong>the</strong> br<strong>in</strong>e is calculated. The calculated SRR is<strong>the</strong>n used to model <strong>the</strong> k<strong>in</strong>etic <strong>isotope</strong> effect on d 18 O SO4 2 .Fig. 8 shows that a fractionation factor <strong>of</strong> 16.25‰ is toosmall to expla<strong>in</strong> <strong>the</strong> observed d 18 O SO4 2 variations. Whilehigher fractionation factors are able to reproduce <strong>the</strong> peakvalue <strong>of</strong> d 18 O SO4 2 ¼ 28:6& at 27.5 mbsf, <strong>the</strong>y fail to expla<strong>in</strong><strong>the</strong> values observed <strong>in</strong> <strong>the</strong> middle <strong>of</strong> <strong>the</strong> pr<strong>of</strong>ile.Increas<strong>in</strong>g <strong>the</strong> fractionation factor to values greater than57‰ would solve <strong>the</strong> discrepancy between 250 and 60 mbsf,but must result <strong>in</strong> much higher d 18 O SO4 2 values between 60and 0 mbsf (see Fig. 8). An <strong>in</strong>crease <strong>of</strong> <strong>the</strong> SRR <strong>in</strong> <strong>the</strong> upper30 mbsf, a possibility we cannot exclude, would also <strong>in</strong>crease<strong>the</strong> peak for <strong>the</strong> 16‰ model, but this scenario wouldstill be unable to expla<strong>in</strong> <strong>the</strong> d 18 O SO4 2 data at 160 mbfs.The only way to expla<strong>in</strong> <strong>the</strong> observed d 18 O SO4 2 values witha k<strong>in</strong>etic model would be a variable fractionation factor.However, <strong>the</strong> required k<strong>in</strong>etic fractionation factors aremuch higher than those reported <strong>in</strong> <strong>the</strong> literature (up to21‰, see Böttcher et al., 2005). We thus conclude that k<strong>in</strong>eticfractionation <strong>of</strong> oxygen <strong>isotope</strong>s dur<strong>in</strong>g microbial <strong>sulfate</strong>reduction is an unlikely explanation for <strong>the</strong> ODP Site1130 data.The d 18 O SO4 2 values may be also be <strong>in</strong>fluenced by oxygen<strong>isotope</strong> exchange reactions. Under typical mar<strong>in</strong>e conditions,oxygen <strong>isotope</strong> exchange rates are slow (10 7 years,Chiba and Sakai, 1985). However, metabolic <strong>in</strong>termediateslike APS or sulfite facilitate oxygen exchange reactions <strong>in</strong>side<strong>the</strong> cytoplasm (Fritz et al., 1989). In this scenario, <strong>the</strong>overall oxygen <strong>isotope</strong> effect depends on <strong>the</strong> volumetric exchangeflux b, on <strong>the</strong> completeness <strong>of</strong> <strong>the</strong> exchange reaction(k), and <strong>the</strong> isotopic equilibration fractionation factor .While we have no means to determ<strong>in</strong>e k from field measurements,data from ODP Site 1130 clearly shows that at30 mbsf, <strong>the</strong> isotopic signature <strong>of</strong> <strong>the</strong> <strong>in</strong>terstitial <strong>water</strong> iswith<strong>in</strong> error similar to <strong>the</strong> empirical equilibrium constantas determ<strong>in</strong>ed by Fritz et al. (1989). This implies that <strong>the</strong>comb<strong>in</strong>ation <strong>of</strong> k and b is large enough to achieve a completeexchange at this depth. We <strong>the</strong>refore assume thatk = 1, and acknowledge that we may underestimate b.Our model was built without a priory assumptionswhich <strong>of</strong> <strong>the</strong> metabolic <strong>in</strong>termediates facilitates <strong>the</strong> oxygen<strong>isotope</strong> exchange, and we are <strong>the</strong>refore unable to differentiatebetween <strong>the</strong> <strong>in</strong>dividual contributions <strong>of</strong> APS and sulfite.The measured d 18 O SO4 2 data reaches values <strong>of</strong> up to 28.6‰,whereas <strong>in</strong> experiments with sulfur disproportionat<strong>in</strong>g bacteria<strong>the</strong> oxygen <strong>isotope</strong> exchange between <strong>water</strong> and sulfiteand subsequent reoxidation to <strong>sulfate</strong> typically results <strong>in</strong>much lower values <strong>of</strong>f up to 21‰ at 28 to 35 °C (Böttcheret al., 2001, 2005). Assum<strong>in</strong>g that <strong>the</strong> oxidation <strong>of</strong> sulfiteto <strong>sulfate</strong> adds one oxygen from <strong>the</strong> cytoplasmic <strong>water</strong>(d 18 O=0‰), <strong>the</strong> <strong>in</strong>itial oxygen <strong>isotope</strong> equilibrium betweensulfite and <strong>water</strong> <strong>in</strong> <strong>the</strong> above experiments wouldhave been close 28.6‰.To achieve <strong>the</strong> observed d 18 O SO4 2 ¼ 28:6& through <strong>the</strong>addition <strong>of</strong> a <strong>water</strong>-derived oxygen requires that ei<strong>the</strong>r <strong>the</strong><strong>in</strong>itial sulfite–<strong>water</strong> equilibrium was 38.1‰ or that <strong>the</strong>added oxygen was enriched relative to <strong>the</strong> ambient <strong>water</strong>.In <strong>the</strong> case <strong>of</strong> <strong>sulfate</strong> reduc<strong>in</strong>g bacteria, <strong>the</strong> most likelysource <strong>of</strong> <strong>the</strong> oxygen used for <strong>the</strong> oxidation <strong>of</strong> sulfite toAPS is adenos<strong>in</strong>e monophosphate (AMP, see Fig. 9).AMP is used <strong>in</strong> <strong>the</strong> transformation <strong>of</strong> sulfite to APS, a reactionthat is reversibly catalyzed by <strong>the</strong> flavoenzyme APSreductase (Fritz et al., 2002). While we do not know <strong>the</strong>d 18 O <strong>of</strong> AMP, its oxygen <strong>isotope</strong> composition is likely similarto <strong>the</strong> oxygen <strong>isotope</strong> equilibrium <strong>isotope</strong> fractionationbetween <strong>in</strong>organic phosphate and <strong>water</strong>. Follow<strong>in</strong>g





