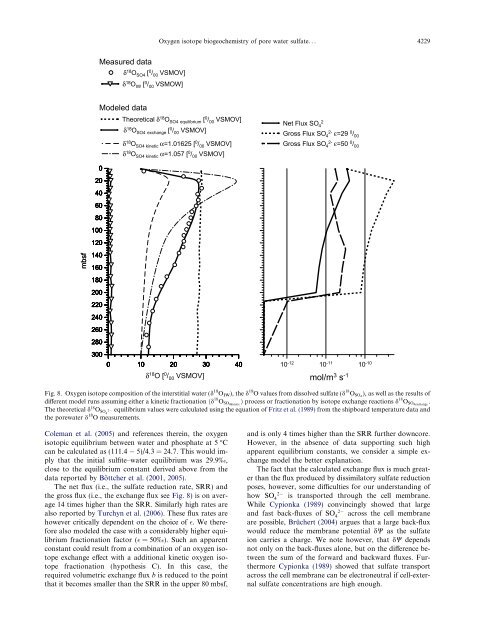
4228 U.G. Wortmann et al. / Geochimica et Cosmochimica Acta 71 (2007) 4221–4232mbsf [m]0255075100125150175200225250275300-50 0 50 100SO 4 [mM] 2-DH 2 S [mM] 2-Dδ 34 S SO 4 [ 0 / 00 ] 2-Dδ 34 S H 2 S [ 0 / 00 ] 2-DΔ 34 S [ 0 / 00 ] 2-DSO 4 [mM] 1-DH 2 S [mM] 1-Dδ 34 S SO 4 [ 0 / 00 ] 1-Dδ 34 S H 2 S [ 0 / 00 ] 1-DΔ 34 S [ 0 / 00 ] 1-DFig. 7. Comparison between <strong>the</strong> results <strong>of</strong> a hypo<strong>the</strong>tical 2-Dmodel with a strong horizontal flow component (l<strong>in</strong>es), and <strong>the</strong>results <strong>of</strong> 1-D model which only considers <strong>the</strong> vertical flowcomponents <strong>of</strong> <strong>the</strong> 2-D model (symbols). The largest differences areseen for <strong>the</strong> concentrations <strong>of</strong> hydrogen sulfite and <strong>sulfate</strong>,suggest<strong>in</strong>g that a 1-D model overestimates biological activity <strong>in</strong><strong>the</strong> presence <strong>of</strong> a lateral flow component. Note however that <strong>the</strong>differences are small, and that <strong>the</strong> difference <strong>in</strong> <strong>the</strong> <strong>isotope</strong> data isnegligible. The above results were obta<strong>in</strong>ed from models which usesimilar horizontal and vertical scales as observed <strong>in</strong> <strong>the</strong> westerntransect <strong>of</strong> ODP Leg 182 (i.e., Sites 1130 and 1132 Feary et al.,2000a). The models used a fractionation factor <strong>of</strong> a = 1.07, <strong>the</strong> 2-Dmodel used horizontal flow velocity <strong>of</strong> x x = 6.867 · 10 11 m/s, andboth models use a vertical flow velocity <strong>of</strong> x z = 3.173 · 10 11 m/s,and a sedimentation rate <strong>of</strong> 7.93 · 10 12 m/s.(Mizutani and Rafter, 1969; Aharon and Fu, 2003). Thefractionation factors for S reported for ODP Site 1130range from 75‰ (direct isotopic difference <strong>of</strong> dissolved2SO 4and H 2 S) to 65‰ (determ<strong>in</strong>ed from reaction transportmodel<strong>in</strong>g, Wortmann et al., 2001). We thus use a m<strong>in</strong>imumfractionation factor <strong>of</strong> 16.25‰, and a maximumfractionation factor <strong>of</strong> 57‰ to <strong>in</strong>vestigate <strong>the</strong> <strong>the</strong>oreticald 18 O SO4 2 evolution with depth if it were caused by a k<strong>in</strong>etic<strong>isotope</strong> effect associated with <strong>the</strong> volumetric <strong>sulfate</strong> reductionrate f. Note that <strong>the</strong> latter fractionation factor ford 18 O SO4 2 is substantially higher than any reported <strong>in</strong> <strong>the</strong> literature(up to 21‰, see Böttcher et al., 2005), and we donot imply that those fractionation factors do exist. Wemerely use this number to explore <strong>the</strong> upper solution envelope<strong>of</strong> <strong>the</strong> model.The volumetric SRR is determ<strong>in</strong>ed from <strong>in</strong>verse model<strong>in</strong>g<strong>of</strong> <strong>the</strong> <strong>sulfate</strong> concentration pr<strong>of</strong>ile (see, Wortmann,2006). The rates shown <strong>in</strong> Fig. 8 are higher than those presentedby Wortmann (2006) as <strong>the</strong> model considers now amajor change <strong>in</strong> <strong>the</strong> sedimentation regime dur<strong>in</strong>g <strong>the</strong> last20 ky (H<strong>in</strong>e et al., 2002) which affects <strong>the</strong> way <strong>the</strong> upwell<strong>in</strong>gvelocity <strong>of</strong> <strong>the</strong> br<strong>in</strong>e is calculated. The calculated SRR is<strong>the</strong>n used to model <strong>the</strong> k<strong>in</strong>etic <strong>isotope</strong> effect on d 18 O SO4 2 .Fig. 8 shows that a fractionation factor <strong>of</strong> 16.25‰ is toosmall to expla<strong>in</strong> <strong>the</strong> observed d 18 O SO4 2 variations. Whilehigher fractionation factors are able to reproduce <strong>the</strong> peakvalue <strong>of</strong> d 18 O SO4 2 ¼ 28:6& at 27.5 mbsf, <strong>the</strong>y fail to expla<strong>in</strong><strong>the</strong> values observed <strong>in</strong> <strong>the</strong> middle <strong>of</strong> <strong>the</strong> pr<strong>of</strong>ile.Increas<strong>in</strong>g <strong>the</strong> fractionation factor to values greater than57‰ would solve <strong>the</strong> discrepancy between 250 and 60 mbsf,but must result <strong>in</strong> much higher d 18 O SO4 2 values between 60and 0 mbsf (see Fig. 8). An <strong>in</strong>crease <strong>of</strong> <strong>the</strong> SRR <strong>in</strong> <strong>the</strong> upper30 mbsf, a possibility we cannot exclude, would also <strong>in</strong>crease<strong>the</strong> peak for <strong>the</strong> 16‰ model, but this scenario wouldstill be unable to expla<strong>in</strong> <strong>the</strong> d 18 O SO4 2 data at 160 mbfs.The only way to expla<strong>in</strong> <strong>the</strong> observed d 18 O SO4 2 values witha k<strong>in</strong>etic model would be a variable fractionation factor.However, <strong>the</strong> required k<strong>in</strong>etic fractionation factors aremuch higher than those reported <strong>in</strong> <strong>the</strong> literature (up to21‰, see Böttcher et al., 2005). We thus conclude that k<strong>in</strong>eticfractionation <strong>of</strong> oxygen <strong>isotope</strong>s dur<strong>in</strong>g microbial <strong>sulfate</strong>reduction is an unlikely explanation for <strong>the</strong> ODP Site1130 data.The d 18 O SO4 2 values may be also be <strong>in</strong>fluenced by oxygen<strong>isotope</strong> exchange reactions. Under typical mar<strong>in</strong>e conditions,oxygen <strong>isotope</strong> exchange rates are slow (10 7 years,Chiba and Sakai, 1985). However, metabolic <strong>in</strong>termediateslike APS or sulfite facilitate oxygen exchange reactions <strong>in</strong>side<strong>the</strong> cytoplasm (Fritz et al., 1989). In this scenario, <strong>the</strong>overall oxygen <strong>isotope</strong> effect depends on <strong>the</strong> volumetric exchangeflux b, on <strong>the</strong> completeness <strong>of</strong> <strong>the</strong> exchange reaction(k), and <strong>the</strong> isotopic equilibration fractionation factor .While we have no means to determ<strong>in</strong>e k from field measurements,data from ODP Site 1130 clearly shows that at30 mbsf, <strong>the</strong> isotopic signature <strong>of</strong> <strong>the</strong> <strong>in</strong>terstitial <strong>water</strong> iswith<strong>in</strong> error similar to <strong>the</strong> empirical equilibrium constantas determ<strong>in</strong>ed by Fritz et al. (1989). This implies that <strong>the</strong>comb<strong>in</strong>ation <strong>of</strong> k and b is large enough to achieve a completeexchange at this depth. We <strong>the</strong>refore assume thatk = 1, and acknowledge that we may underestimate b.Our model was built without a priory assumptionswhich <strong>of</strong> <strong>the</strong> metabolic <strong>in</strong>termediates facilitates <strong>the</strong> oxygen<strong>isotope</strong> exchange, and we are <strong>the</strong>refore unable to differentiatebetween <strong>the</strong> <strong>in</strong>dividual contributions <strong>of</strong> APS and sulfite.The measured d 18 O SO4 2 data reaches values <strong>of</strong> up to 28.6‰,whereas <strong>in</strong> experiments with sulfur disproportionat<strong>in</strong>g bacteria<strong>the</strong> oxygen <strong>isotope</strong> exchange between <strong>water</strong> and sulfiteand subsequent reoxidation to <strong>sulfate</strong> typically results <strong>in</strong>much lower values <strong>of</strong>f up to 21‰ at 28 to 35 °C (Böttcheret al., 2001, 2005). Assum<strong>in</strong>g that <strong>the</strong> oxidation <strong>of</strong> sulfiteto <strong>sulfate</strong> adds one oxygen from <strong>the</strong> cytoplasmic <strong>water</strong>(d 18 O=0‰), <strong>the</strong> <strong>in</strong>itial oxygen <strong>isotope</strong> equilibrium betweensulfite and <strong>water</strong> <strong>in</strong> <strong>the</strong> above experiments wouldhave been close 28.6‰.To achieve <strong>the</strong> observed d 18 O SO4 2 ¼ 28:6& through <strong>the</strong>addition <strong>of</strong> a <strong>water</strong>-derived oxygen requires that ei<strong>the</strong>r <strong>the</strong><strong>in</strong>itial sulfite–<strong>water</strong> equilibrium was 38.1‰ or that <strong>the</strong>added oxygen was enriched relative to <strong>the</strong> ambient <strong>water</strong>.In <strong>the</strong> case <strong>of</strong> <strong>sulfate</strong> reduc<strong>in</strong>g bacteria, <strong>the</strong> most likelysource <strong>of</strong> <strong>the</strong> oxygen used for <strong>the</strong> oxidation <strong>of</strong> sulfite toAPS is adenos<strong>in</strong>e monophosphate (AMP, see Fig. 9).AMP is used <strong>in</strong> <strong>the</strong> transformation <strong>of</strong> sulfite to APS, a reactionthat is reversibly catalyzed by <strong>the</strong> flavoenzyme APSreductase (Fritz et al., 2002). While we do not know <strong>the</strong>d 18 O <strong>of</strong> AMP, its oxygen <strong>isotope</strong> composition is likely similarto <strong>the</strong> oxygen <strong>isotope</strong> equilibrium <strong>isotope</strong> fractionationbetween <strong>in</strong>organic phosphate and <strong>water</strong>. Follow<strong>in</strong>g
<strong>Oxygen</strong> <strong>isotope</strong> <strong>biogeochemistry</strong> <strong>of</strong> <strong>pore</strong> <strong>water</strong> <strong>sulfate</strong>... 4229Measured dataδ 18 O SO4 [ 0 / 00 VSMOV]δ 18 O IW [ 0 / 00 VSMOW]Modeled dataTheoretical δ 18 O SO4 equilibrium [ 0 / 00 VSMOV]δ 18 O SO4 exchange [ 0 / 00 VSMOV]δ 18 O SO4 k<strong>in</strong>etic α=1.01625 [ 0 / 00 VSMOV]δ 18 O SO4 k<strong>in</strong>etic α=1.057 [ 0 / 00 VSMOV]Net Flux SO 42Gross Flux SO 4 2- ε=29 0 / 00Gross Flux SO 4 2- ε=50 0 / 00020406080100mbsf1201401601802002202402602803000 10 20 30 40δ 18 O [ 0 / 00 VSMOV]10 -12 10 -11 10 -10mol/m 3 s -1Fig. 8. <strong>Oxygen</strong> <strong>isotope</strong> composition <strong>of</strong> <strong>the</strong> <strong>in</strong>terstitial <strong>water</strong> (d 18 O IW ), <strong>the</strong> d 18 O values from dissolved <strong>sulfate</strong> (d 18 O SO4 ), as well as <strong>the</strong> results <strong>of</strong>different model runs assum<strong>in</strong>g ei<strong>the</strong>r a k<strong>in</strong>etic fractionation ðd 18 O SO4k<strong>in</strong>etic Þ process or fractionation by <strong>isotope</strong> exchange reactions d 18 O SO4exchange .The <strong>the</strong>oretical d 18 O SO4 2 equilibrium values were calculated us<strong>in</strong>g <strong>the</strong> equation <strong>of</strong> Fritz et al. (1989) from <strong>the</strong> shipboard temperature data and<strong>the</strong> <strong>pore</strong><strong>water</strong> d 18 O measurements.Coleman et al. (2005) and references <strong>the</strong>re<strong>in</strong>, <strong>the</strong> oxygenisotopic equilibrium between <strong>water</strong> and phosphate at 5 °Ccan be calculated as (111.4 5)/4.3 = 24.7. This would implythat <strong>the</strong> <strong>in</strong>itial sulfite–<strong>water</strong> equilibrium was 29.9‰,close to <strong>the</strong> equilibrium constant derived above from <strong>the</strong>data reported by Böttcher et al. (2001, 2005).The net flux (i.e., <strong>the</strong> <strong>sulfate</strong> reduction rate, SRR) and<strong>the</strong> gross flux (i.e., <strong>the</strong> exchange flux see Fig. 8) is on average14 times higher than <strong>the</strong> SRR. Similarly high rates arealso reported by Turchyn et al. (2006). These flux rates arehowever critically dependent on <strong>the</strong> choice <strong>of</strong> . We <strong>the</strong>reforealso modeled <strong>the</strong> case with a considerably higher equilibriumfractionation factor ( =50‰). Such an apparentconstant could result from a comb<strong>in</strong>ation <strong>of</strong> an oxygen <strong>isotope</strong>exchange effect with a additional k<strong>in</strong>etic oxygen <strong>isotope</strong>fractionation (hypo<strong>the</strong>sis C). In this case, <strong>the</strong>required volumetric exchange flux b is reduced to <strong>the</strong> po<strong>in</strong>tthat it becomes smaller than <strong>the</strong> SRR <strong>in</strong> <strong>the</strong> upper 80 mbsf,and is only 4 times higher than <strong>the</strong> SRR fur<strong>the</strong>r downcore.However, <strong>in</strong> <strong>the</strong> absence <strong>of</strong> data support<strong>in</strong>g such highapparent equilibrium constants, we consider a simple exchangemodel <strong>the</strong> better explanation.The fact that <strong>the</strong> calculated exchange flux is much greaterthan <strong>the</strong> flux produced by dissimilatory <strong>sulfate</strong> reductionposes, however, some difficulties for our understand<strong>in</strong>g <strong>of</strong>2how SO 4is transported through <strong>the</strong> cell membrane.While Cypionka (1989) conv<strong>in</strong>c<strong>in</strong>gly showed that large2and fast back-fluxes <strong>of</strong> SO 4across <strong>the</strong> cell membraneare possible, Brüchert (2004) argues that a large back-fluxwould reduce <strong>the</strong> membrane potential dW as <strong>the</strong> <strong>sulfate</strong>ion carries a charge. We note however, that dW dependsnot only on <strong>the</strong> back-fluxes alone, but on <strong>the</strong> difference between<strong>the</strong> sum <strong>of</strong> <strong>the</strong> forward and backward fluxes. Fur<strong>the</strong>rmoreCypionka (1989) showed that <strong>sulfate</strong> transportacross <strong>the</strong> cell membrane can be electroneutral if cell-external<strong>sulfate</strong> concentrations are high enough.