

Netherlands Journal
NJCC Volume 10, Oktober 2006
NJCC Volume 10, Oktober 2006
- No tags were found...

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
PKC<br />
netherlands journal of critical care<br />
ETC<br />
Mitochondria<br />
PKC<br />
MAPK<br />
A. Contraction<br />
mitoK + ATP<br />
K +<br />
HSP<br />
Mitoc hondria<br />
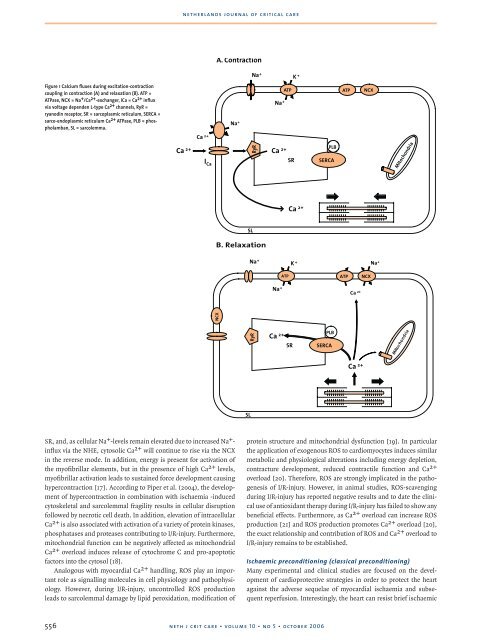
Figure 1 Calcium fluxes during excitation-contraction<br />
coupling in contraction (A) and relaxation (B). ATP =<br />
ATPase, NCX = Na + /Ca 2+ -exchanger, ICa = Ca 2+ influx<br />
via voltage dependen L-type Ca 2+ channels, RyR =<br />
ryanodin receptor, SR = sarcoplasmic reticulum, SERCA =<br />
sarco-endoplasmic reticulum Ca 2+ ATPase, PLB = phospholamban,<br />
SL = sarcolemma.<br />
Ca 2+<br />
SL<br />
Na+ K +<br />
ATP<br />
Na +<br />
•reduced Ca 2+ - overload<br />
•reduced oxidative stress<br />
Na +<br />
•preserved mitochondrial function<br />
Ca 2+<br />
RyR<br />
Ca 2+ Ca 2+<br />
I SR<br />
Ca<br />
PLB<br />
SERCA<br />
ATP NCX<br />
Cardioprotection<br />
•Less necrosis<br />
•Less apoptosis<br />
•Improved contractile recovery<br />
Mitochondria<br />
SL<br />
B. Relaxation<br />
Na+ Na +<br />
ATP<br />
K +<br />
ATP<br />
Na +<br />
NCX<br />
Ca 2+<br />
RyR<br />
NCX<br />
SR<br />
PLB<br />
Ca 2+ Ca 2+<br />
SERCA<br />
Mitoc hondria<br />
SL<br />
SR, and, as cellular Na + -levels remain elevated due to increased Na + -<br />
influx via the NHE, cytosolic Ca 2+ will continue to rise via the NCX<br />
in the reverse mode. In addition, energy is present for activation of<br />
the myofibrillar elements, but in the presence of high Ca 2+ levels,<br />
myofibrillar activation leads to sustained force development causing<br />
hypercontraction [17]. According to Piper et al. (2004), the development<br />
of hypercontraction in combination with ischaemia -induced<br />
cytoskeletal and sarcolemmal fragility results in cellular disruption<br />
followed by necrotic cell death. In addition, elevation of intracellular<br />
Ca 2+ is also associated with activation of a variety of protein kinases,<br />
phosphatases and proteases contributing to I/R-injury. Furthermore,<br />
mitochondrial function can be negatively affected as mitochondrial<br />
Ca 2+ overload induces release of cytochrome C and pro-apoptotic<br />
factors into the cytosol [18].<br />
Analogous with myocardial Ca 2+ handling, ROS play an important<br />
role as signalling molecules in cell physiology and pathophysiology.<br />
However, during I/R-injury, uncontrolled ROS production<br />
leads to sarcolemmal damage by lipid peroxidation, modification of<br />
protein structure and mitochondrial dysfunction [19]. In particular<br />
the application of exogenous ROS to cardiomyocytes induces similar<br />
metabolic and physiological alterations including energy depletion,<br />
contracture development, reduced contractile function and Ca 2+<br />
overload [20]. Therefore, ROS are strongly implicated in the pathogenesis<br />
of I/R-injury. However, in animal studies, ROS-scavenging<br />
during I/R-injury has reported negative results and to date the clinical<br />
use of antioxidant therapy during I/R-injury has failed to show any<br />
beneficial effects. Furthermore, as Ca 2+ overload can increase ROS<br />
production [21] and ROS production promotes Ca 2+ overload [20],<br />
the exact relationship and contribution of ROS and Ca 2+ overload to<br />
I/R-injury remains to be established.<br />
Ischaemic preconditioning (classical preconditioning)<br />
Many experimental and clinical studies are focused on the development<br />
of cardioprotective strategies in order to protect the heart<br />
against the adverse sequelae of myocardial ischaemia and subsequent<br />
reperfusion. Interestingly, the heart can resist brief ischaemic<br />
556<br />
neth j crit care • volume 10 • no 5 • october 2006




