THE AMERICAN SOCIETY OF HEMATOLOGY - hematology.fi
THE AMERICAN SOCIETY OF HEMATOLOGY - hematology.fi
THE AMERICAN SOCIETY OF HEMATOLOGY - hematology.fi

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
THe AmericAn SocieTy of HemATology<br />
ASH 2011<br />
AnnuAl Meeting 10.–13.12.2011, sAn diego, cAliforniA, usA
ASH 2011 -kongressiraportti<br />
Julkaisija: novartis <strong>fi</strong>nland oy, metsänneidonkuja 10, 02130 espoo. www.novartis.<strong>fi</strong> Puhelin 09-6133 200<br />
Päätoimittajat: professori ja ylilääkäri Kimmo Porkka, Biomedicum Helsinki ja HyKS hematologian klinikka ja<br />
kliinisen hematologian dosentti Tarja-Terttu Pelliniemi, Turun yliopisto | Toimitusneuvosto: Kimmo Porkka, Tarja-<br />
Terttu Pelliniemi, meri Kekäle ja Terhi Taipale-lepola. | Toimitus ja ulkoasu: miltton oy | Paino: lönnberg Painot oy<br />
Valokuvat: mikael lindén, Shutterstock: kansi (Star of india, San Diego)
sisällys:<br />
Kimmo Porkka & Tarja-Terttu Pelliniemi//<br />
Pääkirjoitus ............................................. s.2<br />
Pekka Anttila//<br />
Multippeli myelooma – hoitovaihtoehdot<br />
tarkentuvat ja lisääntyvät .......................... s.4<br />
Kimmo Porkka//<br />
KMl – Parantava hoito näköpiirissä .......... s.12<br />
Minna lehto//<br />
Myelo<strong>fi</strong>broosi – parantavaa lääkehoitoa<br />
saadaan vielä odottaa hetkinen ................ s.18<br />
Veli Kairisto//<br />
Molekyyligenetiikan uusilla työkaluilla<br />
tehoa täsmädiagnostiikkaan ja<br />
jäännöstaudin monitorointiin .................. s.24<br />
Mika Kontro//<br />
AMl:n kohdennetut hoidot tulossa ........... s.34<br />
Tero Pirttinen//<br />
Myelodysplastiset oireyhtymät –<br />
uutta tietoa perimän muutoksista<br />
ja hoitovaihtoehdoista............................. s.38<br />
Ulla Pihkala//<br />
Virustorjuntaohjelma<br />
kantasolujensiirtopotilaille ..................... s.42<br />
Vesa lindström//<br />
Krooninen lymfaattinen leukemia (Kll) –<br />
vihdoinkin pintaa syvemmälle? ................ s.48<br />
ASH 2011<br />
1
Kimmo Porkka & Tarja-Terttu Pelliniemi:<br />
PääKirjoiTUs<br />
Amerikan hematologiyhdistyksen (AsH) järjestyksessään<br />
53. vuosikokous järjestettiin<br />
san Diegossa, Kalifornian auringon alla.<br />
osallistujia lämmitti myös kokouksen sähköinen<br />
ja innostunut ilmapiiri alan uusimpien<br />
aikaansaannosten ääreltä. Perinteen<br />
mukaan esityksiä oli potilasvuoteelta perustutkimuslaboratorioon,<br />
veritautien koko kirjo<br />
kattaen. Dr. johnsonia mukaillen ”When a<br />
man is tired of AsH, he is tired of <strong>hematology</strong>”.<br />
Väsyttävää taasen oli Atlantin yli reissaaminen,<br />
ja pian kokouksen jälkeen julkaistavat<br />
webcastit (<strong>hematology</strong>.org, maksullisia)<br />
keskeisimmistä luennoista vain lisäävät<br />
tulevina vuosina houkutusta jäädä viettämään<br />
joulunalusaikaa koti-suomeen, netin<br />
kautta AsH-annista nauttien.<br />
Kuten olettaa sopi, oli genomiikka<br />
uuden polven sekvensointiteknologian myötä<br />
vahvasti esillä, kuten myös sen mukanaan<br />
tuomat uudet työvälineet niin diagnostiikkaan,<br />
hoitovasteen arviointiin kuin hoitojen<br />
kehitykseenkin. Verisyöpien lisäksi koko genomin<br />
tai eksomin sekvensointia hyödynnettiin<br />
muun muassa tukosalttiuden selvittämiseen<br />
ja allogeenisen verenkantasolujen luovuttajan<br />
ja saajan perimien vertailuun. Tämä<br />
genomiikan esiinmarssi hematologisten tautien<br />
diagnostiikkaan ja hoitoon näkyy hyvin<br />
myös tämän raportin kirjoituksista. sama<br />
kehitys tulee jatkumaan ja jopa korostumaan<br />
myös seuraavissa AsH-kokouksissa.<br />
onkin aivan kutkuttavaa seurata kuinka<br />
monen yleisen veritaudin patogeneesi alkaa<br />
pikkuhiljaa avautua, tässä kokouksessa esimerkkeinä<br />
olivat AMl, MDs ja Kll. Tämän<br />
vuoden aikana olemme lisäksi saaneet tietoa<br />
myös plasmasolutautien ja lymfoomien<br />
geneettisestä patogeneesista. Hieman<br />
yllättävä löydös on ollut somaattisten geenimuutosten<br />
suhteellisen pieni lukumäärä,<br />
useimmiten alle 10 mutaatiota/potilas, joista<br />
taudin syntyyn osallistuvien poikkeavuuksien<br />
(driver mutations) seulonta kuulostaa<br />
hyvinkin mahdolliselta lähivuosien aikana.<br />
Haasteena on yhä klonaalisen diversiteetin<br />
ja evoluution ymmärtäminen, johon kuitenkin<br />
kehittynyt sekvensointiteknologia antaa<br />
aivan uusia mahdollisuuksia.<br />
syövän patogeneesin kannalta ei näytä<br />
olevan suurta väliä sillä, missä kudoksessa<br />
tai solutyypissä geenipoikkeamat todetaan.<br />
Esimerkiksi taudin aiheuttava BrAF-mutaatio<br />
voi yhtä hyvin olla melanooma- kuin karvasoluleukemiasolussa,<br />
AlK-mutaatio keuhkosyövässä<br />
ja lymfoomissa, tai KiT-mutaatio<br />
gliomiissa, AMl:ssa tai GisT-kasvaimessa.<br />
lisäksi on löydetty uusia tähän saakka lähes<br />
tuntemattomia syntymekanismeja, kuten<br />
kokouksessa esitellyt rNA:n silmukointiin<br />
liittyvät sF3B1-mutaatiot niinkin erilaisissa<br />
taudeissa kuin MDs:ssa (N Engl j Med<br />
2011;364:2496–506) ja Kll:ssa (painossa,<br />
12.12.2011 NEjM.org). Varsinaiset mekanismit<br />
ovat vasta avautumassa ja opittavaa<br />
on paljon. Vaikuttaa kuitenkin ilmeiseltä, että<br />
lähivuosien aikana leukemioiden ja muidenkin<br />
syöpien luokitus tulee (jälleen) muuttu-
maan ja perustana käytetään yksilökohtaista<br />
avainmutaatiopro<strong>fi</strong>ilia kasvaimen sijainnista<br />
riippumatta. Ehkäpä tämä tulee näkymään<br />
jopa sairaalatasolla; 2021 meillä saattaa<br />
olla vaikkapa hypermetylaatiotautien klinikka,<br />
jossa hoidetaan samoin perussäännöin<br />
laajaa ryhmää ulkoasultaan aivan erilaisia<br />
syöpätauteja. Tässäpä haastetta niin lääkärikouluttajille<br />
kuin hallintonikkareillekin.<br />
Entistä tärkeämpää on potilasnäytteiden<br />
systemaattinen varastointi biopankkiin<br />
niin tutkimuksellista kuin kliinistäkin käyttöä<br />
varten. on aivan selvää, että tarve palata<br />
potilaan tautihistorian aikana kerättyihin<br />
näytteisiin on suuri tiedon ja hoitojen kehittyessä.<br />
sHy:n ryhdyttyä ennakkoluulottomasti<br />
verisolupankkiiriksi on meillä tähän<br />
kansainvälisestikin poikkeuksellisen hyvät<br />
edellytykset käynnistyneen FHrB-hankkeen<br />
myötä (<strong>hematology</strong>.<strong>fi</strong>/fhrb).<br />
AsH-kokouksen kerma oli koottu plenary-sessioon,<br />
joka tänä vuonna oli poikkeuksellisen<br />
tasokas. siinä esiteltiin yksi harvoista<br />
satunnaistetuista allogeeniseen kantasolujensiirtoon<br />
liittyvistä tutkimuksista<br />
(UsA), jossa tutkittiin siirteen lähteen (veri<br />
vs. luuydin) vaikutusta ja todettiin, että eroa<br />
potilaiden elossaolossa ei ollut, mutta käänteishyljintää<br />
veren siirrettä käytettäessä oli<br />
hieman enemmän. lienee hyvä jäädä odottamaan<br />
julkaisua paperimuodossa ennen<br />
sen kummempia johtopäätelmiä. japanilaiset<br />
tutkijat esittelivät iPs-peräistä immortalisoitua<br />
megakaryosyyttisolulinjaa, jota<br />
voitaisiin mahdollisesti hyödyntää lähitulevaisuudessa<br />
verihiutaleiden tuottamisessa<br />
in vitro. Hemo<strong>fi</strong>lian geenihoito otti myös<br />
merkittävän askeleen eteenpäin, ainakin jos<br />
kysytään plenary-luennon innostuneen esittäjän<br />
mielipidettä.<br />
George Daley (Boston) piti E. Donnel<br />
Thomas -palkintoluennon, joka oli kokouksen<br />
yksi hienoimmista esityksistä. oli koskettavaa<br />
kuulla yhden henkilön kehitystarina<br />
alkaen varhaisista lääketieteen opinnoista –<br />
johtaen hauskojen sattumien kautta – uraa<br />
uurtaviin löydöksiin niin leukemogeneesin<br />
(Ph-positiiviset taudit) kuin kantasolujenkin<br />
aloilla. Esityksessä huomioitiin nöyrästi<br />
ohjaajien ja mentoreiden keskeinen osuus<br />
kuten myös suuren yhteistyöverkoston, jota<br />
ilman mitään ei voi saada aikaan.<br />
Eräs merkittävä kokouksessa esitetty<br />
julkistus oli Bob löwenbergin (Erasmusyliopisto,<br />
rotterdam) nimitys Blood-lehden<br />
ensimmäiseksi ei-amerikkalaiseksi päätoi-<br />
ASH 2011<br />
mittajaksi. Ehkäpä tämäkin on yksi osoitus<br />
kasvavasta kilpailusta ja eurooppalaisen<br />
hematologian aseman vahvistumisesta. Toki<br />
EHA-kokouksella on vielä matkaa etenkin<br />
tutkimuspuolen tarjonnan osalta AsH-kokoukseen,<br />
eikä italialaisvetoinen Haematology<br />
vielä vedä vertoja Blood-lehden kattavuudelle,<br />
mutta erot ovat koko ajan pienenemässä<br />
ja ala kansainvälistymässä. on selvää, että<br />
seuraava laajentumiskohde on Aasia, jossa<br />
jo nyt tehdään merkittävä osa hematologisesta<br />
perustutkimuksesta (esimerkkinä<br />
vaikkapa Pekingin valtaisat koko genomin<br />
sekvensointi<strong>fi</strong>rmat).<br />
suomalaista väriä nähtiin jälleen niin<br />
puhujapöntöissä kuin posterimeren laineillakin.<br />
Petri Bono esitteli hienosti AsH/<br />
AsCo-symposiumissa imatinibiylläpitohoidon<br />
eduista GisT-kasvainten hoidossa, ja<br />
muun muassa sirpa lepän ja Esa jantusen<br />
työt olivat hyvin esillä. Ei nyt aivan pienellä<br />
ilolla ja ylpeydellä mainitsisin myös oman<br />
ryhmämme kaksi suullista ja neljä posterimuodossa<br />
ollutta esitystä, jotka ovat vuosien<br />
työn ja osaavien tutkijaystäviemme<br />
yhteistyön aikaansaannoksia.<br />
Tässä AsH 2011 -julkaisussa esitellään<br />
aiempaan tapaan kirjoittajien omakohtaisia<br />
näkemyksiä kokouksen huippukohdista.<br />
olemme koonneet raporttiin myelooman,<br />
Kll:n, MDs:n, myelo<strong>fi</strong>broosin ja KMl:n<br />
ympärillä olleita esityksiä. lisäksi voitte<br />
lukea pediatrian ja laboratoriolääketieteen<br />
AsH-helmistä kirjoittajien oman näkemyksen<br />
höystäminä.<br />
Kirjoituksissa viitteet on merkitty abstraktinumerolla,<br />
joita vastaavat tekstit löytyvät<br />
osoitteesta bloodjournal.org. sähköinen<br />
versio tästä ja aiemmista AsH-raporteista<br />
on sHy-ohjesivuilla (<strong>hematology</strong>.<strong>fi</strong>/<br />
ash), linkki iPad-versioon on lähetetty sähköpostitse<br />
tai sitä voi tiedustella osoitteesta<br />
kysymys.palaute@novartis.com.<br />
suurkiitos Tarja-Terttu Pelliniemelle<br />
artikkeleiden taitavasta editoinnista ja<br />
Novartisille AsH-raportin tukemisesta.<br />
Antoisia lukuhetkiä!<br />
Kimmo Porkka<br />
Professori, ylilääkäri<br />
HyKs:n hematologian klinikka ja<br />
Hematologinen tutkimusyksikkö<br />
3
Pekka Anttila, HYKS:<br />
MUlTiPPEli MyElooMA – HoiToVAiHToEHDoT<br />
TArKENTUVAT jA lisääNTyVäT
Myelooma on viime vuosina ollut yksi AsH-kokouksen keskeisistä teemoista.<br />
san Diegossa asiaa käsiteltiin yhdessä koulutusohjelmassa ja myeloomalle<br />
oli varattu suullisia esityksiä varten melkoinen määrä istuntoja.<br />
Myelooma-hakusanalla löytyy viittaus 773 tutkimukseen. Voitaneen todeta,<br />
että tänä vuonna ei ollut tarjolla yhtään läpimurtoluontoista suuren<br />
satunnaistetun tutkimuksen ensiesittelyä. Tämän vuoden tärkeimpänä<br />
antina pitäisin jo pidempään käynnissä olleiden tutkimuksien päivityksiä,<br />
selvästi lisääntymässä olevien ylläpitohoitoa koskevien tutkimuksien<br />
tuloksia sekä uusia lääkeaineita koskevia tutkimuksia hoidolle refraktaarissa<br />
ja uusiutuneessa myeloomassa.<br />
SYTogeneTiiKKA<br />
HoiToPääTöSTen TuKenA<br />
leif Bergsagel (Mayo Clinic) piti koulutusohjelmassa<br />
erinomaisen luennon myelooman<br />
sytogenetiikasta otsikolla ”Many multiple<br />
myelomas: making more of the molecular<br />
mayhem”. Tavattoman selkeään ja kantaa<br />
ottavaan esitykseen kannattaa tutustua<br />
myös Hematology-julkaisun sivuilla<br />
(http://asheducationbook.<strong>hematology</strong>library.org/).<br />
Erityisenä kiinnostuksen kohteena<br />
on ollut t(4;14) (p16;q32) -translokaatio,<br />
joka aikaansaa fuusiogeenin MMsET-FGFr3.<br />
Tämän fuusion osuutta myeloomageneesissa<br />
ei vielä täysin tunneta. MMsET-fuusiopartneri<br />
ilmentyy kaikissa t(4;14)-tuumoreissa, kun<br />
taas FGFr3:n ilmentymä voi puuttua (25 %).<br />
Villityyppi-FGFr3 osallistunee myös taudin<br />
syntyyn etenkin sen varhaisvaiheessa ja siksi<br />
FGFr3 sopii kohteeksi kohdennetulle lääkehoidolle.<br />
FGFr3-proteiinin ilmentymä voidaan<br />
osoittaa virtaussytometrilla.<br />
Kliinikon mielenkiinto kohdistuu sytomolekyyligenetiikan<br />
tutkimustulosten merkitykseen<br />
hoitovalinnoissa. Bergsagel esitteli<br />
tässä yhteydessä päivitetyn version Mayoklinikan<br />
riskiryhmän mukaisesta msMArThoitokaaviosta<br />
(kuva 1).<br />
siinä suuren riskin ryhmään kuuluvat<br />
ne potilaat, joilla todetaan del17p, t(14;16),<br />
t(14;20) tai mikrosirututkimuksessa havaitaan<br />
suuren riskin geeni-ilmentymäpro<strong>fi</strong>ili.<br />
Keskiriskiin luetaan t(4;14), G-raitatutki-<br />
ASH AsH 2011<br />
muksessa nähty del13, hypodiploidi karyotyyppi<br />
tai jos Pli (plasmasolujen jakaantumisindeksi)<br />
on suurempi kuin 3 %. Tavanomaiseen<br />
riskiin kuuluvat t(11;14), t(6;14),<br />
hyperdiploidi kromosomisto ja kaikki muut<br />
muutokset. FisH-tutkimukset tulisi tehdä<br />
selekoiduista plasmasoluista.<br />
Erityisesti t(4;14) -keskiriskin muutoksen<br />
tunnistaminen diagnoosivaiheessa<br />
on tärkeää, koska tämä ryhmä hyötyy erityisesti<br />
riittävän pitkästä bortetsomibi-hoidosta<br />
(vähintään vuoden ajan). suuren riskin<br />
ryhmässä mikään yksittäinen lääkeaine ei<br />
paranna ennustetta. Kliinisten tutkimusten<br />
ulkopuolella suuren riskin potilaille suositellaan<br />
VrD-hoitoja (bortetsomibi-lenalidomidi-deksametasoni)<br />
vähintään vuoden ajan<br />
(+ autologisella kantasolusiirteellä tuettu<br />
intensiivihoito neljän hoidon jälkeen). Tavanomaisen<br />
riskin ryhmässä on suosituksena<br />
neljä bortetsomibipohjaista hoitoa ja intensiivihoito<br />
(autologinen kantasolujensiirto).<br />
uuTTA TieToA SATunnAiSTeTuiSTA<br />
HoiToTuTKimuKSiSTA<br />
jesus san Miquel (salamanca) esitteli päivityksen<br />
VisTA-tutkimuksesta (#476). Tutkimuksessa<br />
verrattiin kokeellista melfalaaniprednisoni-bortetsomibihoitoa<br />
(VMP) melfalaani-prednisoni<br />
-hoitoon (MP). lääkehoito<br />
kesti molemmissa haaroissa 54 viikkoa.<br />
Tutkimukseen satunnaistettiin 682 potilasta<br />
(344 VMP-haaraan ja 338 MP-haaraan).<br />
5
6<br />
Kuva 1. Mayo-KliniKan risKiryhMän MuKainen MsMarT-hoiToKaavio<br />
FisH<br />
MUUT<br />
siirToKElPoisET<br />
siirTooN<br />
KElPAAMATToMAT<br />
Matala riski<br />
t(11;14)<br />
t(6;14)<br />
Hyperdiploidi<br />
Muut<br />
4 sykliä rd tai<br />
CyBorD<br />
Autologinen<br />
kantasolujen<br />
siirto<br />
Kerää<br />
kantasolut<br />
jatka rD<br />
rd tai MPT<br />
seuranta<br />
Keskiriski<br />
t(4;14)<br />
sytogeneettinen<br />
deleetio 13 tai<br />
hyperdiploidi tai....<br />
4 sykliä CyBorD<br />
Autologinen<br />
kantasolujen siirto<br />
Bortezomibi<br />
pohjainen hoito<br />
vähintään<br />
vuoden ajan<br />
MP + viikottainen<br />
Bortezomibi tai<br />
viikottain CyBorD<br />
Bortezomibi ylläpito<br />
ASH 2011<br />
Korkea riski<br />
del17p<br />
t(14;16)<br />
t(14,20)<br />
GEP korkea riski<br />
4 sykliä Vrd<br />
Autologinen<br />
kantasolujen siirto,<br />
erityisesti jos<br />
ei täydellisessä<br />
remissiossa<br />
Vrd vähintään<br />
yhden vuoden ajan<br />
Vrd
Mediaaniseuranta-aika oli 60,1 kuukautta.<br />
Vain 5 % potilaista on hävinnyt seurannasta.<br />
Tässäkin analyysissä jo aiemmin osoitettu<br />
elinaikaetu kokeellisen hoidon hyväksi<br />
säilyi. Mediaanielinaika oli 56,4 kuukautta<br />
VMP-haarassa ja 43,1 kuukautta MP-haarassa.<br />
Elinaikaetu nähtiin useimmissa alaryhmissä.<br />
Tutkimuksessa oli vain 46 potilasta,<br />
joilla oli todettu epäsuotuisa sytogeneettinen<br />
pro<strong>fi</strong>ili. Tässä ryhmässä ei elinaikaetua<br />
havaittu. sekundaarimaligniteeteissa ei<br />
havaittu lisääntymistä syöpätautien taustaesiintymiseen<br />
verrattuna.<br />
Gareth Morgan (royal Marsden) esitteli<br />
suuresta MrC iX -tutkimuksesta tällä kerralla<br />
talidomidiylläpitohoitoa koskevia tuloksia<br />
(#993). Tutkimukseen oli satunnaistettu<br />
kaikkiaan 1970 potilasta. Mukana olivat erikseen<br />
omina ryhminään intensiivihoidon saaneet<br />
potilaat ja tavanomaisen lääkehoidon<br />
saaneet potilaat. jo aikaisemmin tästä tutkimuksesta<br />
on julkaistu tulokset tsoledronihapon<br />
vaikutuksesta elinaikaennusteeseen. Tutkimuksen<br />
mukaan tsoledronihappo lisää odotettavissa<br />
olevaa elinaikaa 5,5 kuukaudella.<br />
Nyt esiteltiin tuloksia talidomidiylläpitohoidosta.<br />
oleellinen uusi tieto tässä tutkimuksessa<br />
on potilaan sytogeneettisen löydöksen<br />
merkitys ennusteeseen. Huonon ennusteen<br />
sytogeneettisiksi löydöksiksi valittiin t(4;14),<br />
t(14;16), t(14;20), 1q+, ja 17p–, (sekä 1p32<br />
- vain intensiivihoitoryhmässä). Nyt esitettiin<br />
tulokset kuuden vuoden seurannasta. Uusiutumista<br />
edeltävä elinaika piteni merkitsevästi<br />
talidomidiylläpitohoitoa saaneilla potilailla<br />
(mediaani 22 vs.16 kuukautta). Hyöty<br />
rajoittui kuitenkin suotuisan sytogeneettisen<br />
pro<strong>fi</strong>ilin omaavien potilaiden ryhmään. Talidomidiylläpitohoito<br />
ei parantanut koko potilasjoukon<br />
elinaikaennustetta ja heikensi sitä<br />
epäsuotuisan sytogenetiikan ryhmässä. Näyttää<br />
siltä, että uusien lääkkeiden käytön optimointi<br />
puoltaa sytomolekyyligeneettisten tutkimusten<br />
tekemistä nykyistä laajemmin myeloomaa<br />
sairastavilla potilailla.<br />
ASH 2011<br />
Antonio Palumbo (Torino) esitteli<br />
satunnaistettua lumekontrolloitua tutkimusta<br />
vanhemmilla myeloomapotilailla, jossa<br />
vertailtiin kolmea ryhmää niin, että kaikki<br />
ryhmät saivat yhdeksän sykliä melfalaanipohjaista<br />
hoitoa. yksi ryhmä sai vain melfalaani-prednisoni<br />
-hoidot (MP), toinen ryhmä<br />
vastaavasti melfalaani-prednisoni-lenalidomidi<br />
-hoidot (MP-r) ja kolmas ryhmä<br />
melfalaani-prednisoni-lenalidomidi- hoitojen<br />
lisäksi vielä lenalidomidi-ylläpitohoidon<br />
(MPr-r) (#475). Potilaita oli yhteensä 459<br />
ja he olivat vähintään 65-vuotiaita. lenali-<br />
”Näyttää siltä, että uusien<br />
lääkkeiden käytön<br />
optimointi puoltaa<br />
sytomolekyyligeneettisten<br />
tutkimusten tekemistä<br />
nykyistä laajemmin<br />
myeloomaa sairastavilla<br />
potilailla.”<br />
domidihoidossa ylläpitohoitolääkkeenä oli<br />
lenalidomidi 10 mg 21 päivän ajan 28 päivän<br />
sykleissä. Mediaaniseuranta-aika oli 30 kuukautta.<br />
MPr-r -hoito vähentää merkittävästi<br />
taudin etenemisen todennäköisyyttä muihin<br />
hoitovaihtoehtoihin verrattuna 65–75-vuotiailla<br />
potilailla. Tällä hetkellä mediaaniselviäminen<br />
taudin etenemiseen on 31 kuukautta,<br />
mikä on huomattavan pitkä. Hematologisia<br />
sekundaarimaligniteettea nähtiin<br />
MPr-r -ryhmässä 3, MPr-ryhmässä 1 ja<br />
MP-ryhmässä ei yhtään. Kaikissa ryhmissä<br />
7
8<br />
nähtiin kaksi kiinteää kasvainta. Kirjoittajat<br />
arvioivat, että hoidosta saatavat hyödyt ylittävät<br />
sekundaarisyöpien tuottaman haitan.<br />
Kolmen vuoden kohdalla MPr-r -ryhmässä<br />
elossa oli 73 % potilaista ja MP-ryhmässä<br />
65 %. Ero ei ole ainakaan vielä merkitsevä<br />
(p = 0,254).<br />
Maria-Victoria Mateos (salamanca)<br />
esitti espanjalaisen ryhmän työn, jossa vertailtiin<br />
iäkkäillä potilailla induktiohoidossa<br />
bortetsomibi-melfalaani-prednisonihoitoa<br />
bortetsomibi-melfalaani-talidomidihoitoon<br />
(#477). Potilaita oli yhteensä 260. Kumpaakin<br />
hoitoa potilaat saivat yhden kuuden viikon<br />
syklin ja viisi viiden viikon sykliä. induktiohoidoissa<br />
hoitotulokset olivat yhteneväiset. Hoitovasteen<br />
(orr) saavutti 80 ja 81 % potilaista.<br />
Tässä vaiheessa potilaat (n=178) satunnaistettiin<br />
kummassakin ryhmässä uudelleen<br />
saamaan bortetsomibi-prednisoni tai bortetsomibi-talidomidi<br />
-ylläpitohoitoa ylläpitohoidossa<br />
potilaat saivat yhden kolmen viikon<br />
syklin bortetsomibia (1,3 mg/m 2 päivinä 1,<br />
4, 8 ja 11) kolmen kuukauden välein ja lisäksi<br />
joko prednisonia 50 mg joka toinen päivä tai<br />
talidomidia 50 mg/pv kolmen vuoden ajan.<br />
ylläpitohoidon aikana hoitovaste syveni ja virtaussytometrilla<br />
mitattu jäännöstaudin määrä<br />
pieneni. jäännöstautivapaiden potilaiden<br />
osuus suureni ylläpitohoidon aikana 24–42<br />
%. Hoidon alusta laskettuna mediaaniaika<br />
ilman taudin uusiutumista oli 35 kuukautta<br />
ja kokonaiselinajan mediaani oli 60 kuukautta.<br />
ylläpitohoidon satunnaistamisesta alkaen<br />
VT-hoidossa olevat potilaat selvisivät 30<br />
kuukautta ilman taudin uusiutumista (hoitojen<br />
alusta lukien 39 kuukautta) ja VP-potilaat<br />
24 kuukautta. VT-ryhmässä nähtiin kuitenkin<br />
merkittävää toksisuutta. 7 %:lla sydäntapahtumia,<br />
11 %:lla gastrointestinaalista toksisuutta<br />
ja 9 %:lla gradus 3–4 neuropatiaa.<br />
(VP-ryhmässä vastaavasti 1 %, 3 % ja 3 %).<br />
Tutkijat ehdottivat, että bortetsomibia tulisi<br />
jatkossa tutkia myös ylläpitohoidossa lenalidomidiin<br />
kombinoituna.<br />
ASH 2011<br />
YlläPiToHoidon ASemA mYeloomAn<br />
HoidoSSA<br />
ylläpitohoitoa koskevia tutkimuksia oli esillä<br />
useita. Koulutusohjelmassa Donna reece<br />
(Toronto) kävi läpi ylläpitohoitotutkimuksia,<br />
joita oli tehty käyttäen talidomidiä, lenalidomidiä<br />
ja bortetsomibia. Talidomidiylläpitohoidosta<br />
on juuri julkaistu meta-analyysi,<br />
jonka mukaan talidomidiylläpito tuottaa<br />
elinaikaetua. lääkkeen käyttö on ylläpitohoidossa<br />
jäänyt kuitenkin vähäiseksi lähinnä<br />
haittavaikutuksien takia. lenalidomidiylläpitohoitoa<br />
on tutkittu sekä Euroopassa<br />
että yhdysvalloissa. Tässä kokouksessa esiteltiin<br />
tuloksia näistä tutkimuksista. Bortetsomibin<br />
käytöstä ylläpitohoidossa esitettiin<br />
elinaikaetua näyttävä tutkimus vuoden 2010<br />
AsH:ssa. (AsH 2010 #40). Asia oli esillä<br />
myös tänä vuonna. NCCN (National Comprehensive<br />
Cancer Network) Guide line on listannut<br />
nämä lääkkeet ylläpitohoidossa näyttöön<br />
perustuvaksi hoidoksi. Muissa Käypä<br />
Hoito -ohjeissa ei juurikaan vielä ole kirjauksia<br />
ylläpitohoidosta vakiintuneena hoitona.<br />
Näillä lääkkeillä ei ole myöskään myyntilupaa<br />
ylläpitohoitoindikaatiolla.<br />
RelAPSoiTuneen mYeloomAn HoiTo –<br />
vAloA Tunnelin PääSSä<br />
relapsien hoito on oleellinen osa myelooman<br />
hoitoa. Eliniän ennusteen paraneminen<br />
edellyttää, että relapsit hoidetaan mahdollisimman<br />
tehokkaasti. Koska nykylääkkein<br />
saavutettavat hoitotulokset ovat riittämättömiä,<br />
kohdistui suuri mielenkiinto<br />
AsH:n vuosikokouksessa uusilla lääkkeillä<br />
tehtyihin tutkimuksiin uusiutuneessa ja refraktaarissa<br />
myeloomassa. Vahvan perustan<br />
teemalle rakensi professori Kenneth Anderson<br />
kokouksen koulutusohjelmassa luennoimalla<br />
aiheesta ”New insights into Therapeutic<br />
Targets in Myeloma”. Koko uusi<br />
lääkekehitys on perustunut yhä parempaan<br />
solunsisäisten viestinvälitysketjujen<br />
ymmärtämiseen. Uusia molekyyleja on luo-
tu kertyneen perustiedon pohjalta. Proteosomin<br />
estoon on tulossa uusia molekyylejä.<br />
immunomoduloivien lääkkeiden ryhmään<br />
saadaan uutta. Histonideasetylaasiestäjät<br />
voivat olla jatkossa tärkeä uusi ryhmä,<br />
samoin mTor-estäjät. lisäksi näkyvissä on<br />
myeloomasoluun kohdistuvia monoklonaalisia<br />
vasta-aineita. on hyvin mahdollista,<br />
että ainakin pomalidomidi, kar<strong>fi</strong>ltsomibi ja<br />
elotutsumabi tulevat saamaan myyntiluvan<br />
lähimpien kahden vuoden kuluessa. Tulevaisuudessa<br />
sopiva lääkehoito voidaan todennäköisesti<br />
valita yksilölllisesti potilaan<br />
myeloomasolujen biologisten ominaisuuksien<br />
perusteella. Toistaiseksi joudutaan vielä<br />
hakemaan mahdollisimman tehokasta<br />
hoitoa ilman potilaskohtaista spesi<strong>fi</strong>ä kohdetta.<br />
Näyttää siltä, että useimmat nyt tutkittavat<br />
uudet lääkkeet eivät tule olemaan<br />
riittävän tehokkaita yksinään käytettyinä.<br />
Käytännössä uudet tutkimukset on tehty<br />
kombinoimalla uusi lääke bortetsomibiin<br />
tai lenalidomidiin. jo aiemmin on julkaistu<br />
erinomaisia tuloksia lenalidomidi-bortetsomibi-deksametasonikombinaatiossauusiutuneen<br />
ja refraktaarin taudin hoidossa.<br />
Pomalidomidi on uusi talidomidijohdannainen<br />
immunomodulatoristen lääkeaineiden<br />
ryhmässä. Xavier leleu esitti ranskalaisia<br />
tuloksia pomalidomidista refraktäärisen<br />
myelooman hoidossa (#812). Pomlidomidia<br />
annosteltiin joko 21/28 pv (ryhmä 1;<br />
43 potilasta) tai 28/28 pv (ryhmä 2; 41 potilasta).<br />
sekä lenalidomidille että bortetsomibille<br />
refraktaareja potilaita oli 74 % ryhmässä<br />
1 ja 78 % ryhmässä 2. yli kuusi hoitolinjaa<br />
saaneita potilaita oli vastaavasti 42 % ja 46<br />
%. Vähintään Pr-vasteen saavutti 35 % ja 34<br />
% potilaista. Mediaani tautivapaa elossaoloaika<br />
oli 6,3 kuukautta kummassakin ryhmässä<br />
ja vasteen kesto 11,4 kuukautta ryhmässä<br />
1 ja 7,9 kuukautta ryhmässä 2, mikä puoltaisi<br />
annostelua, jossa pomalidomidia oli 4<br />
mg 21 vuorokautta ja deksametasonia 40 mg<br />
kerran viikossa 28 vuorokauden sykleissä.<br />
ASH 2011<br />
Pomalidomidista esitettiin myös useita<br />
mielenkiintoisia postereita. joseph r. Mikhaelin<br />
työssä (#2942) hoidettiin pomalidomidilla<br />
60 relapsoitunutta potilasta. Mediaani<br />
seuranta-aika oli 30,1 kuukautta. Hoitovasteen<br />
saavutti 65 % potilaista. Vasteen<br />
kesto hoitovasteen saaneilla oli 21,3 kuukautta.<br />
Mediaani selviäminen ilman taudin<br />
etenemistä oli 13 kuukautta ja kahden<br />
vuoden kohdalla elossa oli 76 % potilaista.<br />
Martha lacyn työssä oli mukana 225 relapsoitunutta<br />
potilasta (#3963). Mediaaniseuranta-aika<br />
oli 12,6 kuukautta. 69 % potilaista<br />
oli elossa ja 30 %:lla tauti ei ollut edennyt.<br />
Uudesta proteosomiestäjästä kar<strong>fi</strong>ltsomibista<br />
esitettiin useita tuloksia. ravi<br />
Vij esitti tulokset faasin ii tutkimuksesta<br />
(#813), jossa kar<strong>fi</strong>ltsomibia oli annettu<br />
relapsoituneille ja/tai refraktaareille myeloomapotilaille.<br />
Nyt näytettiin tulokset vain<br />
siitä ryhmästä (67 potilasta), joka ei aikaisemmin<br />
ollut saanut bortetsomibia. Kar<strong>fi</strong>ltsomibia<br />
annosteltiin 20 mg/mE2 lyhyinä<br />
infuusioina pävinä 1, 2, 8, 9, 15, ja 16<br />
28 päivän sykleissä. jatkossa annos suurennettiin<br />
ad 27 mg/m 2 . Kaikkiaan 35 potilasta<br />
saavutti vähintään Pr-vasteen (52 %). Tutkimuksen<br />
seuranta on toistaiseksi niin lyhyt,<br />
että vasteiden mediaanikestoa ei ole saavutettu.<br />
lääke oli hyvin siedetty ja siihen liittyi<br />
vain vähän neuropatiaa.<br />
Elotutsumabi on humanisoitu monoklonaalinen<br />
igG1 vasta-aine, joka kohdistuu<br />
solun pinnan glykoproteiiniin Cs1:een.<br />
Cs1:tä on runsaasti yli 95 %:lla myeloomapotilaista<br />
kasvainsolujen pinnassa. sagar<br />
lonial esitti faasin ii tutkimuksen (#303),<br />
jossa elotutsumabia oli annettu yhdessä<br />
lenalidomidin ja deksametasonin kanssa<br />
relapsoitunutta/refraktaaria multippelia<br />
myeloomaa sairastaville potilaille. Mukana<br />
oli 73 potilasta, jotka olivat saaneet 1–3<br />
aiempaa hoitoa. 55 potilasta oli saanut<br />
aiemmin vähintään kaksi hoitolinjaa. Potilaat<br />
saivat elotutsumabia 28 päivän sykleis-<br />
9
10<br />
sä 10 tai 20 mg i.v. päivinä 1, 8, 15, ja 22<br />
sykleissä 1 ja 2 ja siitä eteenpäin päivinä 1 ja<br />
15. Faasin iii tutkimuksen annokseksi valittiin<br />
10 mg. lenalidomidiannos oli 25 mg päivinä<br />
1–21 ja deksametasonia kerran viikossa<br />
40 mg p.o. paitsi elotutsumabipäivinä 28<br />
mg p.o. ja 8 mg i.v. Elotutsumabin yhteydessä<br />
potilaat saivat esilääkkeinä deksametasonia<br />
8 mg i.v., difenhydramiinia 25–50 mg i.v.<br />
tai p.o., ranitidiinia tai vastaavaa 50 mg i.v.<br />
sekä parasetamolia 500–1000 mg p.o. 92 %<br />
potilaista saavutti hoitovasteen. 22 potilaal-<br />
”Koko uusi lääkekehitys<br />
on perustunut yhä<br />
parempaan solunsisäisten<br />
viestinvälitysketjujen<br />
ymmärtämiseen.”<br />
la tauti oli edennyt, kun mediaaniseurantaaika<br />
oli 11,4 kuukautta. Elotutsumabi-infuusioihin<br />
liittyi reaktioita, mutta nämä oli hallittavissa<br />
esilääkityksen avulla.<br />
Bortetsomibiä pidetään yleisesti sopivana<br />
myelooman peruslääkkeenä, johon voidaan<br />
lisätä erilaisia tutkittavia lääkemolekyylejä.<br />
Paul richardson esitti tuloksia faasin<br />
ii tutkimuksesta, jossa bortetsomibideksametasoni-kombinaatioon<br />
oli liitetty<br />
histonideasetylaasi-inhibiittori panobinostaatti<br />
(#814). Kysymyksessä oli relapsoitunutta/refraktaaria<br />
myeloomaa sairastavat<br />
potilaat. Kahdeksan kolmen viikon syklissä<br />
bortetsomibiannos oli 1,3 mg/m 2 päivinä<br />
1, 4, 8 ja 11, deksametasoni 20 mg bortetsomibipäivinä<br />
ja sitä seuranneina päivinä<br />
ASH 2011<br />
sekä panobinostaatti /lume p.o. päivinä 1,<br />
3, 5, 8, 10 ja 12. Näitä syklejä seurasi neljä<br />
kuuden viikon sykliä, joissa bortetsomibia<br />
ja deksametasonia annosteltiin päivinä 1, 8,<br />
22 ja 29,Panobinostaatti jatkui kolmen viikon<br />
syklien ohjelman mukaan. Potilaat olivat<br />
saaneet keskimäärin neljä aiempaa hoitolinjaa<br />
(2–14). Tutkimuksessa oli mukana<br />
53 potilasta. 44 potilaan tietojen perusteella<br />
oli mahdollista arvioida vastetta. Näistä<br />
yhdeksän saavutti vähintään Pr-vasteen.<br />
Gradus 3–4 haittavaikutuksia oli varsin runsaasti:<br />
trombosytopenia 38 %:lla, uupumus<br />
10 %:lla ja pneumonia 10 %:lla. Perifeerisen<br />
neuropatian määrä oli vain 17 % ja nämä<br />
kaikki olivat grade 1–2.<br />
Paul richardson esitteli myös tuloksia<br />
faasi i/ii -tutkimuksesta, jossa bortetsomibialustalle<br />
liitettiin perifosiini /lume (#815).<br />
Bortetsomibin annokset olivat tavanomaiset.<br />
Kolmen viikon sykleissä bortetsomibi<br />
1,3 mg/m 2 päivinä 1, 4, 8 ja 11, Deksametasoni<br />
20 mg p.o. bortetsomibipäivinä ja lisäksi<br />
seuraavana päivänä etenevässä taudissa.<br />
Perifosiiniannos oli 50 mg kerran päivässä.<br />
Tutkimuksessa oli mukana 84 potilasta. Potilaat<br />
olivat saaneet keskimäärin viisi aiempaa<br />
hoitolinjaa. 73 potilaasta 22 % saavutti<br />
vähintään Pr-vasteen. Bortetsomibirefraktääreillä<br />
potilailla kokonaiselinaika oli keskimäärin<br />
22,5 kuukautta ja bortetsomibin jälkeen<br />
relapsoituneilla vastaavasti 30,4 kuukautta.<br />
Faasin iii tutkimus on meneillään.<br />
Bendamustiini on jo vanhempi molekyyli,<br />
jonka on osoitettu olevan tehokas myeloomassa.<br />
suzanne lentzsch esitti faasin i/<br />
ii tutkimuksen bendamustiini-lenalidomidi-deksametasonikombinaatiostarefraktaarissä<br />
tai relapsoituneessa myeloomassa<br />
(#304). Tutkimuksessa haettiin suurinta siedettyä<br />
annosta. Tutkimuksessa päädyttiin<br />
annokseen bendamustiini 75 mg/m 2 i.v. päivinä<br />
1–2, lenalidomidi 10 mg p.o. päivinä<br />
1–21 sekä deksametasoni päivinä 1, 8, 15<br />
ja 22. Potilaat olivat saaneet aiemmin keski-
määrin kolme hoitoa. Potilaiden mediaaniikä<br />
oli 63 vuotta (38–80). Vähintään Pr-vasteen<br />
saavutti 52 % (13) potilasta. Mediaaniaika<br />
ilman taudin etenemistä oli 4,4 kuukautta.<br />
Kokonaiselinajan mediaania ei vielä<br />
ollut saavutettu. Esittäjä totesi, että hoito on<br />
hyvin siedettyä, mutta suositteli <strong>fi</strong>lgrastiimin<br />
käyttöä. Hyviä vasteita saavutettiin myös<br />
lenalidomidille refraktääreillä potilailla.<br />
james Berenson esitti posterin, jossa<br />
selvitettiin faasi i/ii -tutkimuksessa bendamustiini-bortetsomibi<br />
-kombinaation tehoa<br />
relapsoituneessa/refraktäärissä myeloomassa<br />
(#1857). Faasin i perusteella annosteluksi<br />
tuli bortetsomibi 1,0 mg/m 2 i.v. päivät<br />
1, 4, 8 ja 11 sekä bendamustiini 90 mg/<br />
m 2 i.v. päivät 1 ja 4, 21 päivän sykleissä. 39<br />
potilaasta 11 saavutti vähintään Pr-vasteen.<br />
orr oli 48,7 % (n=19). 70 %:lla potilaista oli<br />
gradus 3–4 haittavaikutuksia, neutropeniaa,<br />
trombosytopeniaa ja anemiaa. Kolme 40:stä<br />
potilaasta kuoli.<br />
selvää on, että jo aivan lähitulevaisuudessa<br />
uusiutuneen multippelin myelooman<br />
hoitoon tullaan saamaan useita tehokkaita<br />
lääkkeitä. Näiden avulla potilaiden elinaikaa<br />
voidaan edelleen pidentää. Potilaiden<br />
hoidon valinta tulee muuttumaan aikaisempaakin<br />
monimutkaisemmaksi. Kovasti kaivattaisiin<br />
myeloomasolujen entistä parempaa<br />
tuntemusta niin, että potilaskohtaisten<br />
biologisten ominaisuuksien perusteella voitaisiin<br />
valita tehokas hoito. Uusia lääkkeitä<br />
on tarjolla ennen kuin tällaisia menetelmiä<br />
on käytettävissä. Uudet hoidot tulevat myös<br />
aiheuttamaan merkittäviä lisäkustannuksia.<br />
Nykyisen tiedon kehittymisen valossa suh-<br />
ASH 2011<br />
11<br />
taudun hyvin optimistisesti myeloomapotilaiden<br />
hoidon kehitykseen. Töitä tulee riittämään,<br />
koska pidentyneen eliniän myötä hoidettavien<br />
potilaiden määrä kasvaa merkittävästi,<br />
ja potilaat tulevat nykytiedon valossa<br />
tarvitsemaan erilaisia hoitoja koko sairauden<br />
ajalle.<br />
loPPuHuiPennuS WAldenTRömin<br />
TAudiSTA<br />
lopuksi esittelen vielä tutkimuksen, joka oli<br />
mukana myös kongressin päättäneen Best of<br />
AsH -ohjelmassa. steven Treonin ryhmä oli<br />
tehnyt kokogenomin sekvensoinnin 30 Waldenströmin<br />
makroglobulinemiaa sairastavan<br />
potilaan lymfoplasmosytoideista soluista<br />
(#300). Parittaisena verrokkina oli tervettä<br />
kudosta kymmeneltä potilaalta. Patologisten<br />
solujen kromosomikohdasta 3p22.2 löytyi<br />
yhden nukleotidin muutos geenissä MyD88<br />
(l265P) 27/30 potilaalta. Mutaatiota ei löytynyt<br />
potilaiden terveistä kudoksista, kahdeksan<br />
myeloomapotilaan CD138-selekoiduista<br />
soluista eikä 12/12 terveeltä verrokilta ja<br />
vain 1/8 igM MGUs potilaalta (jolla tila eteni<br />
nopeasti Waldenströmin makroglobulinemiaksi).<br />
johtopäätöksenä todettiin, että suurelta<br />
osalta Waldenströmin makroglobulinemiaa<br />
sairastavista potilaista löytyy MyD88 l265P<br />
-variantti, jolla funktionaalisten tutkimusten<br />
perusteella oli onkogeenistä aktiviteettia.<br />
Mutaation avulla voidaan erottaa Waldenströmin<br />
makroglobulinemia multippelista myeloomasta<br />
ja igM MGUs-tilanteesta. Aika näyttää,<br />
voidaanko tämän löydöksen avulla löytää<br />
jatkossa myös kohdennettuja hoitoja Waldenströmin<br />
makroglobulinemiaan.
Kimmo Porkka, HYKS:<br />
KMl – PArANTAVA HoiTo NäKöPiirissä
Krooninen myelooinen leukemia (KMl) on ollut syöväntutkimuksen mallitauti<br />
vuosikymmeniä. se on ensimmäinen pahanlaatuinen tauti, johon<br />
liitettiin toistuva geneettinen poikkeavuus (poikkeava kromosomi 22 eli<br />
Philadephia-kromosomi). Tämän geenipoikkeavuuden vuosikymmenten<br />
saatossa kertynyt molekyylitason ymmärrys johti ensimmäiseen tehokkaaseen<br />
täsmähoitoon –BCr-ABl1-tyrosiinikinaasiestäjä (TKE) –joka on<br />
syrjäyttänyt kaikki aiemmat KMl-hoidot. sama malli on tänä päivänäkin<br />
syöpälääkkeiden kehityksen kivijalka: pahanlaatuiseen solukkoon liittyvien<br />
genomin rakennemuutosten kautta päästään nykyisin melko nopeasti<br />
funktionaaliseen tietoon ja sitä kautta voidaan systemaattisesti etsiä täsmätyökaluja<br />
syövän kasvun estämiseen. Teknologian huiman kehityksen<br />
ansiosta ei mene enää kuin muutama vuosi, niin tiedämme kaikki yleisimmät<br />
geenimuutokset, jotka ovat pahanlaatuisten veritautien aiheuttajina.<br />
onkin lupa odottaa jatkoa KMl-tyyppisille tuhkimotarinoille, ainakin<br />
alkuun koskien kroonisia, patogeneesiltaan yksinkertaisempia tauteja.<br />
KMl oli AsH 2011 -kokouksessa näkyvästi<br />
esillä myös siksi, että Ernest Beutler -juhlaluennon<br />
ja -palkinnon jakoivat janet rowley,<br />
Ph-translokaation karakterisoija, ja<br />
Brian Druker, TKE-hoidon pioneeri. Etenkin<br />
86-vuotiaan professori rowleyn huumoria,<br />
tiedettä ja elämänviisautta sisältänyt esitys<br />
oli todella sykähdyttävä.<br />
Voisi ajatella, että KMl on tänä päivänä<br />
ainakin lääkäritutkijoiden mielestä melko<br />
ikävystyttävä sairaus: valtaosa potilaista saa<br />
erinomaisen vasteen suun kautta otettavalle<br />
lääkitykselle, johon liittyy melko vähän haittavaikutuksia.<br />
Vielä kuitenkin riittää pähkinöitä<br />
pureksittavaksi myös tässä taudissa ja<br />
etenkin parantavan lääkehoidon kehityksessä<br />
KMl:n toivotaan pystyvän näyttävän esimerkkiä<br />
pitkälle tulevaisuuteen.<br />
ovATKo uudeT lääKKeeT PARemPiA?<br />
imatinibi (Glivec) on tuttu, tehokas ja turvallinen<br />
lääke, joka on erinomainen ensilinjan hoito<br />
KMl-taudissa. jos jotain voisi toivoa, niin<br />
lisätehoa tarvittaisiin niille, onneksi melko<br />
harvoille, potilaille (5–10 %), joilla tauti etenee<br />
ensimmäisten hoitovuosien aikana. Vaik-<br />
ASH AsH 2011<br />
13<br />
ka imatinibi onkin hyvin siedetty, on vuosikausien<br />
jatkuvan hoidon aikana myös lievemmillä<br />
haittavaikutuksilla merkitystä (turvotukset,<br />
lihaskrampit). joskus kaipaisi myös<br />
hieman ripeämmin tulevaa hoitovastetta.<br />
Näihin haasteisiin pyritään vastaamaan<br />
2. polven TKE-lääkkeillä, joista dasatinibi ja<br />
nilotinibi ovat jo saaneet myyntiluvan ensilinjan<br />
hoitoaiheella, bosutinibi lienee pian<br />
tulossa perässä. Ainoastaan nilotinibilla on<br />
tällä hetkellä erityiskorvattavuus suomessa.<br />
AsH 2011 -kokouksessa esitettiin kolmen<br />
vuoden seuranta-ajan sisältävä päivitys<br />
ENEsTnd-tutkimuksesta, jossa verrattiin<br />
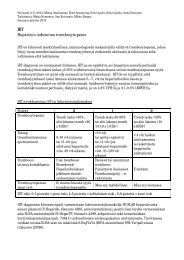
nilotinibia (kaksi annostasoa, 600 ja 800<br />
mg/vrk) imatinibihoitoon (#452, kuva 1).<br />
Esitys vahvisti käsitystä siitä, että nilotinibilla<br />
saavutetaan hoitovaste nopeammin<br />
kuin imatinibilla ja näyttäisi siltä, että saavutettu<br />
etu tautitaakan pienenemisessä (mm.<br />
merkittävä molekylaarinen vaste – MMr)<br />
pysyy myös pidemmän seuranta-ajan jälkeen,<br />
vaikka ero hieman pienenikin.<br />
KMl-potilaille ja heitä hoitaville lääkäreille<br />
esityksen tärkein sanoma oli se, että<br />
myös kolmen vuoden seurannan jälkeen
14<br />
Kuva 1. enesTnd-TuTKiMuKsen raKenne<br />
• N = 846<br />
• 217 KEsKUsTA<br />
• 35 MAATA<br />
PoTilAiDEN lKM<br />
2<br />
rANDoMoiNTi<br />
3<br />
12<br />
NiloTiNiBi 300 MG X 2 (N = 282)<br />
NiloTiNiBi 400 MG X 2 (N = 281)<br />
iMATiNiBi 400 MG X 1 (N = 283)<br />
* strati<strong>fi</strong>oitu sokal-riskiryhmän mukaisesti<br />
ASH 2011<br />
sEUrANTA-AiKA 5 VUoTTA<br />
Kuva 2. Taudin eTeneMinen aKseleraaTiovaiheeseen/blasTiKriisiin<br />
enesTnd-TuTKiMuKsessa 36 KK aiKana<br />
20<br />
15<br />
10<br />
5<br />
0<br />
p = 0,0059<br />
p = 0,0185<br />
2<br />
p = 0,0003<br />
5<br />
p = 0,0085<br />
NiloTiNiBi 300 MG X2 NiloTiNiBi 400 MG X2 iMATiNiBi 400 MG X1<br />
17<br />
sisältää klonaalisen evoluution
nilotinibilla hoidetuilla potilailla on pienempi<br />
riski taudin etenemiseen kuin imatinibihoidetuilla<br />
ja ero oli sekä tilastollisesti merkitsevä<br />
että kliinisesti merkittävä(kuva 2).<br />
samansuuntainen tulos on saatu vastaavanlaisessa<br />
DAsisioN-tutkimuksen (dasatinibi<br />
vs. imatinibi, EHA2011) päivityksessä, kuten<br />
myös BElA-tutkimuksen (bosutinibi vs. imatinibi)<br />
tilannekatsauksessa (#455).<br />
Tulisiko täten kaikille vastatodetuille<br />
KMl-potilaille aloittaa 2. polven TKE-hoito,<br />
koska vaste tulee nopeammin ja progressioita<br />
on vähemmän imatinibiin verrattuna? Näin<br />
saattaa ajan kanssa käydäkin, mutta vielä<br />
kaipaisi muutaman vuoden lisäseurantaa siedettävyyden<br />
ja turvallisuuden suhteen, vaikka<br />
oletusarvona onkin, että suurta eroa ei tule<br />
olemaan. Koska suurimmalle osalle potilaista<br />
imatinibi on riittävän tehokas, olisi kovin<br />
hyödyllistä löytää taudin progressiota ennustavia<br />
tekijöitä. joseph sokalin vuonna 1984<br />
(Blood 1984; 63: 789–799) laskema riskipisteytys<br />
ensilinjan solunsalpaajahoitoa saaneilla<br />
KMl-potilailla pitää melko hyvin kutinsa<br />
myös TKE-aikakaudella, joskin tarvetta on<br />
täsmällisemmille, paremmin taudin biologiaan<br />
perustuville riskitekijöille (ks. alla).<br />
KMl:n päivitetyt hoito-ohjeet löytyvät<br />
osoitteessa http://www.<strong>hematology</strong>.<strong>fi</strong>/cml,<br />
joissa suositellaan harkitsemaan 2. polven<br />
TKE-hoitoa etenkin niille vastatodetuille<br />
KMl-potilaille, joilla on suuren sokal-riskin<br />
tauti. lisäksi hoidon vaihtamista tulisi herkästi<br />
harkita niillä potilailla, joilla on merkittävä<br />
jäännöstauti kolmen kuukauden imatinibihoidon<br />
jälkeen (B-BCr-Qr >10 % isasteikolla)(ks.<br />
#783, #785).<br />
KoHdennuS Kml-KAnTASoluiHin<br />
lääkehoidon kohdennus pahanlaatuisiin<br />
kantasoluihin on ollut aiheena suosittu vuosien<br />
ajan ja ajatuksena on se, että syövän<br />
parantaminen vaatii taudin itujen hävittämistä<br />
tai hallintaa. Kantasoluihin spesi<strong>fi</strong>sti<br />
kohdentuvia lääkkeitä ei ole monia ja yksi<br />
ASH 2011<br />
15<br />
hieman lupausta herättävä ryhmä on ns.<br />
Hedgehog-signaalitiehen vaikuttavat lääkkeet,<br />
kuten smoothened-estäjät (#424).<br />
Aiheeseen tarttui myös pohjoismainen<br />
KMl-ryhmä (NCMlsG) perustuen in vitro<br />
tehtyyn havaintoon siitä, että 2. polven<br />
TKE-lääkityksellä voisi olla tehoa myös kantasolutasolla,<br />
mitä ominaisuutta imatinibilla<br />
ei tunnetusti ole. Metodologisesti kunniahimoinen<br />
NorDCMl006-tutkimus pyrki<br />
arvioimaan saadaanko 2. polven TKE:lla<br />
(dasatinibi) syvempi vaste KMl-kantasoluihin<br />
(Ph+CD34+CD38+/-) kuin imatinibilla ja<br />
lopulliset tulokset esiteltiin suullisessa esityksessä<br />
(#784). Tutkimukseen osallistui 46<br />
vastatodettua KMl-potilasta, joilta kantasoluvasteen<br />
arvioimiseksi kerättiin melko suuri<br />
määrä luuydintä (n. 30 ml), joista keskitetysti<br />
ja standardoidusti laskettiin leukemiakantasolujen<br />
määrä 1 kk, 3 kk ja 6 kk hoidon aloituksesta.<br />
yllättävin ja aiempaan tietoon nähden<br />
ristiriitaisin tulos oli se, että Ph-positiiviset<br />
kantasolut hävisivät luuytimestä nopeasti<br />
hoidon aloituksen jälkeen, jopa nopeammin<br />
kuin kypsemmät mononukleaariset solut.<br />
Eli toisin kuin in vitro, TKE-lääkkeillä on selvä<br />
vaikutus KMl-kantasoluihin in vivo. Tämä<br />
selittänee sen, että osa potilaista voi lopettaa<br />
TKE-lääkityksen, mikäli hoitovaste on ollut<br />
poikkeuksellisen hyvä (stabiili täysi molekylaarinen<br />
vaste, CMr; #603, #604).<br />
NorDCMl006-tutkimus osoitti, että<br />
leukemiakantasolutaakka (Ph+CD34+<br />
CD38neg-solujen osuus) diagnoosihetkellä<br />
on potilaasta toiseen hyvin vaihteleva (1–99<br />
%; kuva 3). leukemiakantasolujen määrä oli<br />
myös vahva ennusteellinen tekijä koskien niin<br />
TKE-hoitovastetta kuin hoidon aiheuttamia<br />
hematologisia haittavaikutuksia: mitä suurempi<br />
oli diagnoosivaiheen leukemiakantasolutaakka,<br />
sitä huonompi oli hoitovaste ja<br />
sitä suurempi todennäköisyys TKE-hoitoon<br />
liittyville sytopenioille. onkin mahdollista,<br />
että KMl-kantasolutaakka kuvaa taudin ikää<br />
eli aikaa siitä, jolloin ensimmäinen Ph-posi-
16<br />
Kuva 3. luuyTiMessä olevien eri KMl-leuKeMiaKanTasolujen<br />
osuus (%) diagnoosivaiheessa<br />
UUsiUTUMisVAPAA ElossAolo (%)<br />
100<br />
80<br />
60<br />
40<br />
20<br />
0<br />
MEDiAANi %<br />
CD34+CD38Pos<br />
N= 43 N= 43 N= 42<br />
95,82<br />
CD34+CD38NEG<br />
ASH 2011<br />
FrAKToiMAToN lUUyDiN<br />
78,79 95,51<br />
p < 0,0001
tiivinen solu syntyi. Vaikka tarkentavia tutkimuksia<br />
tarvitaan, vaikuttaa ilmeiseltä, että<br />
KMl-kantasolujen osuus diagnoosivaiheessa<br />
on uusi taudin perusbiologiaan liittyvä vahva<br />
ennustetekijä. Mikäli menetelmä saadaan<br />
yksinkertaisemmaksi esimerkiksi uusien virtaussytometrisesti<br />
määritettävien KMl-kantasolumerkkiaineiden<br />
avulla (mm. il1rap,<br />
PNAs. 2010;107(37):16280-5), voisi siitä tulla<br />
käyttökelpoinen valittaessa suuren progressioriskin<br />
potilaita tehokkaampaan lääkehoitoon<br />
(2.–3. polven TKE, allogeeninen siirto).<br />
uuSiA lääKKeiTä<br />
Pienelle osalle (
18<br />
minna lehto, HYKS:<br />
MyEloFiBroosi – PArANTAVAA lääKEHoiToA<br />
sAADAAN ViElä oDoTTAA HETKiNEN<br />
ASH 2011
Myeloproliferatiivisten tautien diagnostiikka ja luokittelu on tarkentunut<br />
jAK2-mutaation löytymisen ja uusimpien WHo2008-kriteerien myötä,<br />
mutta arkiset ongelmat potilaiden hoidossa säilyvät. Ketä hoitaa ja miten?<br />
Myelo<strong>fi</strong>broosi (MF) monimuotoisuudessaan on erityisen haasteellinen tauti.<br />
Ainoa parantava hoitokeino on allogeeninen kantasolujensiirto, mutta<br />
se on käytännössä vain pienelle osalle potilaista realistinen hoitovaihtoehto.<br />
Hoitopäätöksiä on ohjannut pyrkimys pienentää tautiin liittyvää riskiä<br />
tromboembolisiin komplikaatiohin sekä transformoitumista akuutiksi<br />
leukemiaksi. lääkkeelliset vaihtoehdot ovat olleet kovin vähäisiä, joten toiveena<br />
on ollut saada kliiniseen käyttöön paitsi hyviä riskiarviointikeinoja<br />
myös uusia lääkevalmisteita. Useammalla lääkeyrityksellä on kliinisissä<br />
tutkimuksissa jAK2-estäjiä, joista ensimmäinen sai myyntiluvan UsA:ssa<br />
AsH 2011 -kokousta edeltävästi. Mutta onko uusista lääkkeistä lupa odottaa<br />
ratkaisua myelo<strong>fi</strong>broosipotilaille?<br />
KouluTuSoSion AnTiA<br />
Vastauksen etsintä oli hyvä aloittaa myeloproliferatiivisten<br />
tautien koulutusosiosta,<br />
jonka viimeisenä ollut Alessandro Vannucchin<br />
luento myelo<strong>fi</strong>broosin hoidosta oli<br />
todella hyvä, tiivis ja mielenkiintoinen kertaus<br />
tämän haasteellisen taudin heterogeenisuudesta.<br />
luokittelujen (iPss/DiPss/<br />
DiPPs plus) avulla potilaat jaetaan riskiryhmiin,<br />
joiden elinaikaennuste vaihtelee<br />
pienen riskin 185 kuukaudesta suuren riskin<br />
16 kuukauteen. luennoitsija kävi läpi<br />
käytössä olevat lääkehoidot sekä splenektomian<br />
ja totesi parhaimmillaankin näiden<br />
perinteisten hoitojen tavoitteeksi elinajan<br />
pidentämisen, mutta mahdollisuuksien<br />
mukaan ainakin tautiin liittyvien oireiden<br />
lieventämisen ja elämänlaadun parantamisen.<br />
Uusista lääkkeistä eniten on odotettu<br />
jAK2-estäjiltä, joista viiden (ruksolitinibi<br />
= iNCB018424, lestaurtinibi= CEP-<br />
701, CyT387, pakritinibi =sB-1518 ja<br />
TG101348=sAr302503) tuloksia luento<br />
lyhyesti sivusi. Vannucchin mukaan on liian<br />
varhaista tehdä johtopäätöksiä jAK2inhibiittoreiden<br />
asemasta MF:n hoidossa.<br />
ASH AsH 2011<br />
19<br />
yksikään näistä lääkkeistä ei ole spesi<strong>fi</strong>nen<br />
jAK2-estäjä vaan vaikuttaa myös muihin<br />
jAK-perheen kinaaseihin (jAK1, jAK3,<br />
Tyk2). ryhmän lääkkeet eroavat toisistaan<br />
niin vaikutuksen kuin haittavaikutuspro<strong>fi</strong>ilinsakin<br />
osalta. Alustavasti lupaavia tuloksia<br />
MF-potilaiden hoidossa on saatu myös<br />
immunomoduloiviin lääkkeisiin kuuluvalla<br />
pomalidomidilla, jolla on parhaillaan<br />
menossa faasi iii -satunnaistettu, lumekontrolloitu<br />
tutkimus verensiirtoriippuvaisten<br />
myelo<strong>fi</strong>broosipotilaiden hoidossa.<br />
Mielenkiintoiselta vaikuttaa lisäksi mTorestäjä<br />
everolimuusi, jonka on in vitro osoitettu<br />
estävän jAK2-mutatoituneiden solujen<br />
proliferaatioita. Nyt odotetaan vahvistuksia<br />
faasi i/ii -tutkimustuloksiin, joiden<br />
mukaan everolimuusi vaikutti niin pernan<br />
kokoon kuin taudin oireisiin ja korjasi anemiaa.<br />
Tutkimuksissa epigeneettisistä lääkkeistä<br />
(HDAC) atsasytidiini ja desitabini olivat<br />
pettymyksiä samoin givinostaatin vaikutus<br />
vain minimaalinen. Vannucchin luennolta<br />
oli tämän kertauksen jälkeen hyvä jatkaa<br />
tiedon hakua kuunnellen oraalisessioiden<br />
antia ja lukien posteririvistöjä.
20<br />
JAK2-eSTäJäT TuovAT<br />
HelPoTuSTA oiReiSiin, muTTA eiväT<br />
PARAnnA TAuTiA<br />
Eniten kokemusta jAK2-inhibiittoreista<br />
on ruksolitinibista, joka sai myyntiluvan<br />
UsA:ssa juuri kokousta edeltävästi. sen on jo<br />
aiemmin tiedetty pienentävän MF-potilaiden<br />
kookasta pernaa ja lieventävän oireita.<br />
Kokouksen aikana sekä CoMForT-i<br />
että CoMForT-ii -tutkimusryhmät julkaisivat<br />
lähinnä alaryhmäanalyysejä. Muista jAK2inbiittoreista<br />
esillä oli pakritinibi (sB1518)<br />
ja faasi i:n osalta mm. ly2784544 (# 2814)<br />
ja sAr302503.<br />
oraalisessiossa MD Anderson Cancer<br />
Centerin srdan Verstovsek kertoi jo aiemmin<br />
julkaistun CoMForT-i -tutkimuksen<br />
alaryhmäanalyysistä (# 278). Tutkimuksen<br />
309 MF-potilaasta 155:llä hoidettiin ruksolitinibilla<br />
15mg x 2 (trombosyytit 100 -200)<br />
tai 20 mg x 2 (trombosyytit >200) ja 154<br />
potilasta lumelääkkeellä. Hoidon vastetta<br />
arvioitiin pernan Mri-kuvauksella (alussa<br />
ja tutkimuksen päättyessä 24 viikon kuluttua<br />
) ja lisäksi seurattiin vastetta oireisiin<br />
potilaiden päivittäin täyttämän oirepäiväkirjan<br />
(Tss total symptom score) avulla. Tutkijat<br />
olivat nyt tehneet alaryhmäanalyysin,<br />
jonka mukaan ruksolitinibi oli lumelääkettä<br />
tehokkaampi pienentämään pernaa ja<br />
lieventämään oireita kaikissa alaryhmissä<br />
(MF, ikä yli /alle 65, iPss-riskiryhmä, jAK2mutaatio,<br />
Hb-taso (yli tai alle 100), palpoituvan<br />
pernan koko tai Tss-pisteet). Esityksensä<br />
lopulla Verstovsek kertoi 13 potilaan<br />
ruksolitinibiryhmästä ja 24 lumeryhmästä<br />
kuolleen ensimmäisen hoitovuoden aikana<br />
(seuranta-aikojen mediaani 52 viikkoa ja<br />
51 viikkoa), mistä tutkijat olivat laskeneet<br />
todennäköisen >48 viikon elossaolon (95<br />
% Ci) 0.98 ruksolitinibiryhmäläisille ja vastaavasti<br />
lumelääkettä saaneilla 0.90, jos Hb<br />
hoidon alkaessa oli ollut yli 100 g/l. Vastaavat<br />
luvut niille, joiden Hb oli alle 100 olivat<br />
0.84 ja 0.77.<br />
ASH 2011<br />
Verstovsekin jälkeen Claire Harrison<br />
puhui tutun CoMForT-ii -tutkimuksen alaryhmäanalyysistä.<br />
Tämän faasi iii monikansallisen<br />
tutkimuksen MF-potilaista 146 hoidettiin<br />
ruksolitinibilla ja 73 hoitavan lääkärin<br />
valinnan mukaan muulla, parhaalla mahdollisella<br />
hoidolla (BAT, best available therapy)<br />
48 viikon ajan. Tulokset pernan pienenemiseen<br />
ja oireiden lievenemiseen on jo aiemmin<br />
julkaistu. Nyt uutta tietoa oli se, että<br />
ruksolitinibi oli BAT:ia tehokkaampi kuin vastetta<br />
arvioitiin pernan pienenemisenä kaikissa<br />
alaryhmissä (ikä, sukupuoli, mutaatiostatus,<br />
iPss riskipisteet, pernan koko hoidon<br />
alkaessa). Varsinaisen tutkimuksen jälkeen<br />
91 (62 %) ruksolitinibiryhmän ja 31 (42 %)<br />
lumeryhmän potilaista jatkaa hoitoaan.<br />
seuraavassa MPN-oraalisessiossa Vertovsek<br />
( #793) kertoi 107 faasi i/ii -tutkimuksissa<br />
ruksolitinibilla hoidetun MF-potilaan<br />
hoitotuloksista verrattuna historialliseen<br />
aineistoon, joka käsitti 310 MDACC, Pavian<br />
ja Milanon sairaaloissa hoidettua MF-potilasta.<br />
ruksolitinibiryhmän potilaista 33 (30,8<br />
%) ja vastaavasti 189 (60,9 %) vertailuaineiston<br />
potilaista kuoli seuranta-ajan ollessa 32<br />
kuukautta ja 22 kuukautta. Monimuuttujaanalyysissa<br />
iPss-keskiriski 2-luokka, vähäisempi<br />
ikä, korkeampi trombosyyttilukema ja<br />
ruksolitinibihoito osoittautuivat itsenäiseksi<br />
tekijöiksi arvioitaessa pidempää elinaikaennustetta.<br />
Ayalew Tefferin työryhmän kokemukset<br />
MF-potilaiden hoidosta ruksolitinibilla<br />
saivat tilaa lauantain posteriseinältä, mutta<br />
eivät yltäneet oraalisessioon. Posteri 1752<br />
kertoi Mayo-klinikan 51 MF-potilaasta, jotka<br />
osallistuivat faasi i/ii -tutkimukseen ruksolitinibilla<br />
lokakuun 2007 ja helmikuun 2009<br />
välisenä aikana. samaisessa tutkimuksessa<br />
oli potilaita myös MD Anderson Cancer<br />
Centerista, joiden tuloksia tämä posteri ei<br />
esitellyt. (MD Andersonin potilaskokemukset,<br />
Verstovsek # 793 ja # 3851). Tulosten<br />
keruun aikana mediaani hoidon aloitukses-
ta oli 3,5 vuotta. ruksolitinibihoidolla perna<br />
pieneni 29 %:lla potilaista, oireet lievenivät<br />
68 %:lla ja ihon kutina vastaavasti 92 %:lla.<br />
Hoitoa jatkaa neljä potilasta, kun 47:n (92<br />
%) hoito oli keskeytynyt. Vuoden kohdalla<br />
hoito oli keskeytynyt 51 %:lla potilaista, kahden<br />
vuoden kuluttua 72 %:lla ja kolmen vuoden<br />
vastaavasti 89 %:lla potilaista. Tavallisin<br />
syy hoidon lopettamiseen oli vasteen puute<br />
tai taudin eteneminen (40 %). Hoitoon liittyvät<br />
haittavaikutukset aiheuttivat noin kolmasosan<br />
hoidonkeskeytyksistä. 18 potilasta<br />
(36 %) oli kuollut ja viidellä (10 %) tauti oli<br />
leukemisoitunut. Vakavia haittavaikutuksia<br />
oli kuudella potilaalla lääkkeen lopettamisen<br />
yhteydessä. Näille tyypillistä oli oireiden<br />
nopea palaaminen, pernan äkillinen ja kivulias<br />
suureneminen, johon liittyi jopa pernainfarkti<br />
sekä akuutti hemodynamiikan romahtaminen.<br />
lääkkeen lopettamisen yhteydessä<br />
nähtiin myös septisen shokin kaltaisia taudinkuvia.<br />
Em. syystä lääkkeen lopettaminen<br />
tulisi tehdä asteittain. Kun potilaita verrattiin<br />
kymmenen vuoden aikana Mayo-klinikassa<br />
hoidettuihin 110-MF -potilaaseen, ei eroa<br />
elossaolossa todettu.<br />
Verstovsek työryhmineen esitti posterissa<br />
(# 3851) faasi i/ii -tutkimukseen osallistuneiden<br />
MD Anderssonissa seurattujen<br />
MF-potilaiden seurantatuloksia. Mediaani<br />
seuranta-ajan ollessa 32 kuukautta, 58/107<br />
(54 %) potilaista oli edelleen hoidossa, 33 oli<br />
kuollut (yhdenkään kuolema ei liittynyt lääkitykseen),<br />
josta tutkijat olivat laskeneet os<br />
69 %. He myös esittivät, että kahden vuoden<br />
elossaolon todennäköisyys oli 3–4 kertainen<br />
niillä, jotka jatkoivat ruksolitinibihoidossa.<br />
rami s. Komrokji (#282) esitteli uuden<br />
jAK2-inhibiittorin, pakritinibin (sB1518)<br />
faasi ii -tutkimuksen tuloksia. ruksolitinibista<br />
poiketen pakritinibi estää tehokkaasti<br />
sekä wild-type jAK2 että jAK2V617F. Tutkimukseen<br />
otettiin 43 MF-potilasta. lääkkeen<br />
annokseksi valikoitui faasi i -tutkimuksesta<br />
400mg/vrk. Hoidon alkaessa mediaa-<br />
ASH 2011<br />
21<br />
ni Hb 102 g/l ja tromb 119 x 10E9/l. Potilaista<br />
15:lla (44 %) trombosyytit olivat alle<br />
100, mukaan lukien seitsemän, joilla luku oli<br />
22<br />
ja kaikki näistä potilaista jatkoivat lääkkeen<br />
käyttöä. 22 (37 %) potilasta on edelleen lääkehoidossa,<br />
hoitosyklien määrän mediaani<br />
28,5 (range 24–41), kun yksi hoitojakso<br />
on 28 vuorokautta. Pernan pienenemisen ja<br />
oireiden lieventymisen lisäksi lääkkeen on<br />
osoitettu vähentävän jAK2V617F-mutatoituneiden<br />
solujen määrää.<br />
Pardanani oli mukana myös tutkimusryhmässä,<br />
joka posterissaan (#3849) esitteli<br />
faasi i/ii -tuloksia jAK1/2-inhibiittorilla<br />
CyT387:lla. 163 potilasta kuudesta keskuksesta<br />
on osallistunut tutkimukseen. lääkkeen<br />
on osoitettu pienentävän pernaa, lieventävän<br />
oireita ja myös korjaavan anemiaa.<br />
lääke on ollut kohtuullisen hyvin siedetty.<br />
Valtaosa haittavaikutuksista on ollut lieviä.<br />
Tavallisin gr 3–4 haittavaikutus oli trombosytopenia<br />
(16 %).<br />
vAi KäännYmmeKö TäSSäKin<br />
dACi (PAn-ACeTYlASe inHibiToR) JA<br />
immunomoduloivien lääKKeiden<br />
Puoleen?<br />
Panobinostaatin (lBH589) faasi i -tutkimuksen<br />
lopullisista tuloksista työryhmän puolesta<br />
oli kertomassa john Mascarenhas (#<br />
794). 18 MF-potilaasta kaikkien oireet lievenivät.<br />
Perna pienentyi ≥ 30 % kymmenellä<br />
potilaalla niistä 11:sta, joilla splenomegalia<br />
oli todettavissa. Viisi potilaista jatkoi<br />
lääkitystä yli 6 kuukautta. Heistä yksi saavutti<br />
16 kuukauden aikana lähes Cr-tasoisen<br />
vasteen eli palpoituva perna pieneni,<br />
veren leukoerytroblastinen verenkuva korjaantui,<br />
Hb nousi 40g/l, minkä lisäksi luuytimessä<br />
nähtiin korjaantumista myös retikuliini/kollageenisäikeiden<br />
vähenemisenä<br />
(gradus 4:stä gradus 1:een 26 hoitosyklin<br />
jälkeen). yksi potilaista tarvitsi viikoittaisia<br />
punasolujensiirtoja Hb-tason pitämiseksi yli<br />
65g/l:n ennen hoidon aloitusta. Puolen vuoden<br />
kuluttua potilaan punasolujensiirtotarve<br />
oli vähentynyt puoleen ja niiden avulla Hbtaso<br />
pysyi 90:ssä.<br />
ASH 2011<br />
lauantain postereissa oli mukana<br />
mm. Begnan, Pardananin, Mesan ja Tefferin<br />
(#1759) yhteenveto pomalidomidilla tehdyistä<br />
faasi i ja ii -tutkimuksista. Pomalidomidi<br />
kuuluu immunomoduloiviin lääkkeisiin<br />
ja sen annos eri tutkimuksissa oli 0,5 mg<br />
tai 2 mg joko ilman tai yhdistettynä kortisoniin.<br />
63/82 (77 %) potilaista tarvitsi tutkimuksen<br />
alkaessa punasolujensiirtoja. Tulokset<br />
kerättiin heinäkuussa 2011 82 potilaasta,<br />
jotka olivat aloittaneet em. tutkimuksissa<br />
toukokuun 2007 ja tammikuun 2010 välisenä<br />
aikana. Pomalidomidilla hoidetuista 27 %<br />
(22/82) anemia koheni. Tämä oli tavallisesti<br />
nähtävissä ensimmäisen kuuden hoitokuukauden<br />
aikana (96 % niistä, joiden anemia<br />
reagoi suotuisasti), mediaani 3,2 kuukautta<br />
(range 1–10 kk). saavutetun vasteen keston<br />
mediaani oli 16,5 kuukautta (2–40). Vaste<br />
lääkkeelle oli todennäköisempi, jos perna ei<br />
ollut massiivinen (jos perna
oli GHVD. Eos (estimated overall survival)<br />
oli viiden vuoden kohdalla 68 % ja kahdeksan<br />
vuoden 65 %. Kolmen vuoden kohdalla<br />
kumulatiivinen relapsi tai taudin eteneminen<br />
oli 22 %:lla. Monimuuttuja-analyysissa<br />
yli 55 vuoden ikä, mismatch-luovuttaja ja<br />
jAK2-mutaation puuttuminen olivat itsenäisen<br />
huonomman ennusteen muuttujia.<br />
loPuKSi<br />
Myelo<strong>fi</strong>broosi on edelleen vaikea vastus.<br />
jAK2-inhibiittoreista ensimmäinen on jo<br />
saanut UsA:ssa myyntiluvan. ruksolitinibi<br />
lieventää useiden potilaiden oireita ja pienentää<br />
pernaa, joten eräs AsH-kongressissa<br />
esiintyneistä ehdotti lääkettä erityisesti MFpotilaille,<br />
joilla on kookas perna ja voimakkaat<br />
tautiin liittyvät oireet. se, miten ruksolitinibin<br />
käyttö kohdentuu, tulee nähtäväksi,<br />
kun se myyntiluvan myötä on useampien<br />
saavutettavissa. jokaisen MF-potilaan lääk-<br />
ASH 2011<br />
23<br />
keeksi siitä ei varmastikaan ole eikä jAK2inhibiittoreilla<br />
ole odotettavissa yhtä hienoa<br />
ja mullistavaa muutosta potilaiden ennusteeseen<br />
kuin aikanaan oli tyrosiinikinaasinestäjien<br />
tullessa KMl-potilaiden hoitoon.<br />
Tutkimuksissa olevat muut jAK2-inhibiittorit<br />
ovat mahdollisesti hematologisilta haittavaikutuksiltaan<br />
vähäisempiä ja joidenkin on<br />
osoitettu vähentävän myös tautitaakkaa.<br />
Mielenkiinnolla jäämme odottamaan<br />
myös pomalidomidin ja panobinostaatin<br />
jatkotuloksia. Väläytettiinpä hiirimalleissa<br />
jo näiden uusien lääkeryhmien yhdistelmähoitoakin.<br />
Näihin on kuitenkin vielä matkaa<br />
ja toistaiseksi käytännön lääkärin on jatkettava<br />
entisin eväin. Vain pieni osa myelo<strong>fi</strong>broosipotilaista<br />
soveltuu allogeeniseen<br />
hoitoon, joten suurin osa jää edelleen hoidettavaksi<br />
nykyisten melko vähäisten lääkevaihtoehtojen,<br />
tukihoitojen ja ison empatian<br />
turvin.
veli Kairisto, TYKSlAb:<br />
MolEKyyliGENETiiKAN UUsillA TyöKAlUillA<br />
TEHoA TäsMäDiAGNosTiiKKAAN jA<br />
jääNNösTAUDiN MoNiToroiNTiiN
Molekyyligenetiikan uusi menetelmä syväsekvenssointi (käytettyjä nimityksiä:<br />
”massively parallel sequencing”, ”next generation sequencing”,<br />
”deep sequencing”) on lyhyessä ajassa mullistanut perustutkimuksen. Tekniikoiden<br />
kehittyminen on ollut nopeaa, laitteistoja on saatavilla jo useilta<br />
valmistajilta ja analyysikustannukset ovat laskeneet nopeasti. on odotettavissa,<br />
että lähitulevaisuudessa tekniikkaa voidaan soveltaa myös kliinisessä<br />
diagnostiikassa. Alan huima kehitys oli nähtävissä AsH 2011 -kongressissa.<br />
syväsekvenssointitekniikkaa oli hyödynnetty useassa sadassa<br />
abstraktissa. Uuden tekniikan ansiosta syöpäsoluihin liittyvät spesi<strong>fi</strong>set ja<br />
toistuvat mutaatiot tullaan kuvaamaan tarkasti jo muutaman lähivuoden<br />
aikana ja on ennakoitavissa, että syöpäsolun genotyyppi tulee olemaan<br />
tärkeä tekijä taudin luokittelussa ja hoitovalinnassa. Eri syöpätyyppien<br />
yhteydessä toistuvasti esiintyvien mutaatioiden tarkka tunnistaminen luo<br />
toivoa myös sille, että tulevaisuudessa löytyy lisää mutatoituneen geenin<br />
toimintaan kohdistuvia täsmälääkkeitä.<br />
Prospektiivista tietoa mutaatioiden kliinisestä<br />
ennusteellisesta merkityksestä kertyy<br />
hitaasti, tosin työssä auttaa se, että monilla<br />
tutkimuskeskuksilla on käytettävissään asianmukaisesti<br />
tutkimusprojektien puitteissa<br />
kerättyä retrospektiivistä näytemateriaalia<br />
(biopankki), jota analysoimalla voidaan saada<br />
nopeasti tietoa eri mutaatioiden kliinisistä<br />
korrelaatioista. Tällaista lähestymistapaa<br />
käyttäneitä tutkimuksia julkaistiin AsH 2011<br />
-kokouksessa useita.<br />
SYväSeKvenSSoinnin SovelTAminen<br />
PAHAnlAATuiSTen veRiTAuTien<br />
JäännöSTAuTiAnAlYTiiKKAAn<br />
Useimmissa kongressissa esitellyissä tutkimuksissa,<br />
missä syväsekvenssointia oli<br />
käytetty jäännöstautianalytiikkaan , lukusyvyys<br />
(alun perin mutatoituneelle geenialueelle<br />
tuotettujen sekvenssien lukumäärä) oli<br />
noin 1000–10 000 päällekkäistä sekvenssiä.<br />
Tällä lukusyvyydellä jäännöstautianalyysin<br />
herkkyys ei vielä kilpaile mutaatiospesi<strong>fi</strong>sen<br />
kvantitatiivisen PCr:n kanssa, jossa saavutetaan<br />
10 –5 –10 –6 :n herkkyystaso. Kuitenkin<br />
muutaman AsH-kongressissa esitellyn tut-<br />
ASH AsH 2011<br />
25<br />
kimuksen perusteella syväsekvenssoinnilla<br />
olisi teknisesti mahdollista saavuttaa kvantitatiiviseen<br />
PCr:ään rinnastettavissa oleva<br />
tai jopa parempi herkkyystaso (#2540,<br />
#2542, #4104).<br />
sekvenssointilukusyvyyden on teknisesti<br />
mahdollista olla jopa useita miljoonia, mutta<br />
samalla on välttämätöntä, että sekvenssoinnin<br />
lukutarkkuus pysyy erittäin hyvänä, sillä muutamakin<br />
virheellisesti luettu emäs miljoonan<br />
oikein luetun joukossa johtaa epäspesi<strong>fi</strong>seen<br />
taustaan, mikä heikentää merkittävästi jäännöstautianalyysissä<br />
saavutettavissa olevaa<br />
herkkyystasoa. Pyrittäessä jäännöstautianalyytikassa<br />
huippuherkkyyteen muodostuu tutkittavan<br />
näytteen määrä rajoittavaksi tekijäksi.<br />
Esimerkiksi kvantitatiivisessa Aso-PCr:ssä<br />
käytetään lähtömateriaalina yleensä 1,5<br />
ug DNA:ta, mikä vastaa noin 240 000 solun<br />
DNA:n määrää. Tällöin teoreettisesti saavutettavissa<br />
oleva maksimiherkkyystaso on 1/240<br />
000 eli 4 x 10 –6 . Pyrittäessä tätä parempaan<br />
herkkyyteen on jäännöstautianalyysiin otettavan<br />
näytteen määrää kasvatettava.<br />
yhden AsH-esityksen perusteella syväsekvenssoinnilla<br />
olisi jäännöstautianalytii-
26<br />
kassa saavutettavissa jopa kvantitatiivista<br />
PCr:ää parempi herkkyystaso. Kyseinen<br />
työ koski kroonisen lymfaattisen leukemian<br />
jäännöstautianalytiikkaa allogeenisen kantasolujensiirron<br />
jälkeen. jäännöstautianalyysin<br />
kohteena käytettiin klonaalista immunoglobuliinin<br />
raskaan ketjun geenin uudelleenjärjestymää.<br />
syväsekvenssointi tehtiin<br />
illumina Hiseq -laitteistolla ja kloonien<br />
tunnistamisessa ja kvanti<strong>fi</strong>oinnissa käytettiin<br />
sequenta inc. -yhtiön tarkoitusta varten<br />
kehittämää bioinformatiikkaohjelmistoa.<br />
jäännöstautianalyysissä veren mononukleaaristen<br />
solujen DNA:ta eristettiin mediaani<br />
2,4 miljoonasta solusta (vaihteluväli<br />
1,1–23,7 milj. solua). jäännöstautianalyysi<br />
tehtiin samanaikaisesti sekä kvantitatiivisella<br />
Aso-PCr:llä että syväsekvenssoinnilla<br />
(#2542). Merkittävältä osin nämä kaksi<br />
metodia antoivat samansuuntaisia tuloksia.<br />
syväsekvenssoinnilla mahdollisesti saavutettavissa<br />
olevaan parempaan herkkyyteen<br />
viittasi kuitenkin se, että 14 potilaan allogeenisen<br />
kantasolusiirron jälkeen otetuista<br />
seurantanäytteistä peräti 43 % (16/37) oli<br />
negatiivisia Aso-PCr:llä, mutta vielä positiivisia<br />
herkkyysrajan (10 –6 ) tuntumassa syväsekvenssoinnilla<br />
tutkittuna. Kahdelle näistä<br />
potilaista kehittyi kliininen relapsi, toiselle jo<br />
38 päivää ja toiselle 644 päivää näytteenottopäivän<br />
jälkeen. Nähtäväksi jää, onko tällainen<br />
tulos toistettavissa myös muiden tutkijoiden<br />
toimesta ja joutuuko kvantitatiivinen<br />
PCr luovuttamaan paikkansa syväsekvenssoinnille<br />
herkimpänä mahdollisena spesi<strong>fi</strong>sen<br />
nukleiinihapposekvenssin kvantitatiivisena<br />
mittausmenetelmänä.<br />
Aml:n moleKYYligeneTiiKKA<br />
DNMT3A-geenin mutaatiot ovat yksi esimerkki<br />
akuuttiin myeloiseen leukemiaan<br />
assosioituvista mutaatioista, joita ei tunnettu<br />
ennen syväsekvenssoinnin käyttöönottoa.<br />
DNMT3A-mutaatio löytyy n. 15 %:lta kaikista<br />
AMl-potilaista, mutta normaalikaryotyypin<br />
potilailta yli 20 %:lta. lisäksi mutaatiota<br />
löytyy merkitsevästi useammin nukleofosmiinigeenin<br />
(NPM1) suhteen mutatoituneilta<br />
kuin mutatoitumattomilta. DNMT3-mutaatio<br />
saattaa olla se tekijä, jonka avulla NPM1mutatoituneiden<br />
AMl-potilaiden joukosta on<br />
tunnistettavissa huonompiennusteinen alaryhmä<br />
(#3519). DNMT3-mutaatiot vaikut-<br />
ASH 2011<br />
taisivat olevan hyvin stabiileja tautisolukloonissa.<br />
Toisessa tutkimuksessa seurattiin 70<br />
potilasta, joilla oli diagnoosivaiheessa tunnistettu<br />
DNMT3-geenin mutaatio. 13 potilasta<br />
relapsoitui, ja kaikilla heillä mutaatio<br />
säilyi myös relapsin aiheuttaneessa kloonissa.<br />
sen sijaan 103 muulle relapsipotilaalle,<br />
joilla tauti oli alun perin DNMT3-mutatoitumaton,<br />
mutaatiota ei ilmaantunut yhdenkään<br />
potilaan relapsiklooniin, vaikka karyotyypin<br />
muutoksia oli todettavissa 39 %:lla<br />
relapsipotilaista. Edellä olevan perusteella<br />
DNMT3-mutaatio soveltuisi erinomaisesti<br />
jäännöstautimarkkeriksi (#409).<br />
NUP98-NsD1 on toinen AMl:aan assosioituva<br />
mutaatio, joka on tunnettu vasta<br />
lyhyen aikaa. Kyseessä on kryptinen fuusiogeeni,<br />
joka ei ole tunnistettavissa kromosomitutkimuksella.<br />
se assosioituu voimakkaasti<br />
huonoon ennusteeseen ja löytyy<br />
hyvin harvoin, n. 2–3 %:lla aikuis-AMl-potilaista,<br />
mutta huomattavasti useammin, n.<br />
15 %:lla, pediatrisista AMl-potilaista. Fuusiogeeniä<br />
ei tavata samanaikaisesti NPM1-<br />
tai CEBPA-mutaatioiden kanssa, joten sitä<br />
kannattaisi seuloa PCr:llä näiltä potilailta.<br />
NUP98-NsD1 -fuusiogeenin tutkiminen<br />
kvantitatiivisella PCr:llä seurantanäytteistä<br />
soveltuu hyvin herkäksi jäännöstautianalyysiksi<br />
(#2531).<br />
rUNX1-geenin mutaatiot ovat osoittautuneet<br />
hyvin yleisiksi AMl-potilailla. Niitä<br />
tavataan noin neljänneksellä kaikista potilaista.<br />
rUNX1-mutaatiot näyttäisivät assosioituvan<br />
saksalaisen tutkimuksen perusteella<br />
resistentimpään tautiin. Hyvin erityyppisiä<br />
rUNX1-geenissä sijaitsevia mutaatioita on<br />
kuvattu, mikä vaikeuttaa mutaatioiden tunnistamista<br />
perinteisillä menetelmillä. syväsekvenssointi<br />
tarjoaa mahdollisuuden kattavaan<br />
rUNX1-geenin mutaatioiden tunnistamiseen<br />
ja mahdollistaa lisäksi mutaatiospesi<strong>fi</strong>sen<br />
herkän jäännöstautianalytiikan (#747).<br />
CEBPA-geenin mutaatioiden osoittamista<br />
on jo usean vuoden ajan pidetty keskeisenä<br />
pienen, n. 10 %:n suuruisen, erittäin<br />
hyväennusteisen AMl:n alaryhmän<br />
osoittamiseksi. CEBPA-mutaatioilla on kliinistä<br />
merkitystä vain, mikäli potilaalla on<br />
kaksi mutaatiota. CEBPA-kaksoismutatoituneet<br />
potilaat näyttävät löytyvän pääsääntöisesti<br />
niiden potilaiden joukosta, joilla ei<br />
ole samanaikaista NPM1-geenin mutaatiota.
siksi kliinisessä diagnostiikassa saattaa olla<br />
tarkoituksenmukaista rajata CEBPA-mutaatioanalyysi<br />
tähän potilasryhmään. Hyvästä<br />
ennusteesta huolimatta osa CEBPA-kaksoismutatoituneista<br />
potilaista relapsoituu, joten<br />
tämän ennakoimiseksi jäännöstautianalytiikka<br />
on tarpeellista myös näiltä potilailta.<br />
CEBPA-mutaatio itsessään on osoittautunut<br />
hyväksi ja stabiiliksi jäännöstautimarkkeriksi.<br />
Valitettavasti CEBPA-mutaatioiden vaihtelevan<br />
luonteen ja koon vuoksi jäännöstautianalyysi<br />
Aso-PCr:llä ei aina ole mahdollista,<br />
mutta syväsekvenssointi näyttäisi tarjoavan<br />
yleisesti käyttökelpoisen ratkaisun<br />
CEBPA-geeniin kohdistuvaan jäännöstautianalytiikkaan<br />
(#2517).<br />
FlT3 ja NPM1-mutaatioiden käytöstä<br />
herkkinä AMl:n jäännöstautimarkkereina on<br />
jo useiden vuosien kokemus myös suomalaisilla<br />
potilailla. NPM1-mutaatiota on pidetty<br />
jäännöstautianalytiikkaan käyttökelpoisempana,<br />
sillä lähes kaikissa tapauksissa diagnoosivaiheessa<br />
todettu NPM1-mutaatio säilyy<br />
myös mahdollisessa relapsikloonissa.<br />
sen sijaan FlT3-pituusmutaation suhteen<br />
tiedetään, että diagnoosivaiheen mutaatio<br />
säilyy vain noin 70–80 %:ssa tapauksista<br />
mahdollisessa relapsissa. NPM ja FlT3mutaatioiden<br />
käyttökelpoisuudelle jäännöstautimarkkereina<br />
saatiin tukea useista<br />
AsH 2011 -kongressissa esitellyistä tutkimustuloksista.<br />
Hematologisessa remissiossa<br />
mitattu NPM1-mutatoituneiden solujen<br />
osuuteen perustuvan jäännöstaudin määrän<br />
osoitettiin saksalaisessa isoon potilasaineistoon<br />
perustuvassa tutkimuksessa hyvin vahvaksi<br />
tautivapaaseen ja kokonaiselinaikaan<br />
korreloivaksi ennustekijäksi (#1445). NPMmutaation<br />
avulla mitattu jäännöstauti allogeenisen<br />
siirron jälkeen osoittautui toisessa<br />
tutkimuksessa käyttökelpoiseksi immunoterapeuttisen<br />
hoidon ohjaajaksi. (#3061).<br />
Vaikka FlT3-mutatoitunutta kloonia<br />
voidaan erittäin herkästi monitoroida kvantitatiivisen<br />
Aso-PCr:n avulla, diagnoosivaiheessa<br />
tutkimuksen käyttökelpoisuutta<br />
rajoittaa se, että vähäisempiä subklooneja<br />
ei pystytä toteamaan ja mutatoituneen valtakloonin<br />
osuus ei ole suoraan mitattavissa.<br />
Valtakloonin osuudella diagnoosivaiheessa<br />
on osoitettu olevan ennusteellista merkitystä<br />
suuremman alleelisen osuuden ja mahdollisen<br />
homotsygotisoitumisen korreloituessa<br />
ASH 2011<br />
27<br />
huonompaan hoitovasteeseen. syväsekvenssoinnin<br />
avulla on periaatteessa mahdollista<br />
tunnistaa diagnoosivaiheessa kaikki mutatoituneet<br />
subkloonit ja jäännöstautiseurannan<br />
kuluessa on mahdollista tunnistaa mahdollinen<br />
alkuperäisen valtakloonin korvautuminen<br />
jollain toisella FlT3-geenin mutaatiolla<br />
(#3548). syväsekvenssoinnistakaan ei<br />
kuitenkaan ole apua siihen, että 20–30 %:n<br />
todennäköisyydellä mahdollinen relapsiklooni<br />
on FlT3-mutaatioiden suhteen kokonaan<br />
negatiivinen. yhdessä abstraktissa<br />
tästä jopa vedettiin johtopäätös, että FlT3pituusmutaatiota<br />
ei tulisi käyttää jäännöstautimarkkerina<br />
(#3557). Useimmissa tapa-<br />
”syväsekvenssointi tarjoaa<br />
mahdollisuuden kattavaan<br />
rUNX1-geenin mutaatioiden<br />
tunnistamiseen ja<br />
mahdollistaa lisäksi<br />
mutaatiospesi<strong>fi</strong>sen herkän<br />
jäännöstautianalytiikan.”<br />
uksissa alkuperäinen mutaatio kuitenkin säilyy<br />
myös relapsivaiheessa ja näissä tapauksissa<br />
mutaatiopositiivisuus edeltää ad 10<br />
kk:lla kliinistä relapsia (#3530). johtopäätöksenä<br />
edellä mainitusta voitaneen pitää,<br />
että FlT3-mutaatiolla voidaan mitata hoitovastetta<br />
hoidon alussa. Myöhemmin mitattuna<br />
vain positiivisella FlT3-löydöksellä on<br />
merkitystä ja negatiivisen löydöksen avulla<br />
ei varmuudella voida poissulkea relapsikloonin<br />
kehittymistä. Mikäli FlT3-mutatoituneelta<br />
potilaalta löytyy diagnoosivaiheessa<br />
jokin toinen stabiilimmaksi tiedetty mutaatio,<br />
kuten NPM1-geenin mutaatio, tulisi<br />
sitä käyttää jäännöstautimarkkerina FlT3mutaation<br />
sijaan.<br />
FlT3-pituusmutaatioon liittyvä mielenkiintoinen<br />
löydös on, että mutatoituneen<br />
alleelin osuuden lisäksi myös mutaatiossa
28<br />
duplikoituneen alueen pituudella näyttäisi<br />
olevan ennusteellista merkitystä. Mutaation<br />
pituus tulee aina selvitettyä, kun FlT3mutaatiota<br />
tutkitaan. Toistaiseksi tätä yksityiskohtaista<br />
tietoa ei kuitenkaan ole sisällytetty<br />
mutaatiolöydöksestä annettuun lausuntoon.<br />
Duplikoituneen alueen pituus voi<br />
vaihdella muutamasta emäksestä aina yli<br />
sataan emäkseen mediaanipituuden ollessa<br />
noin 45 emästä. jaettaessa potilaat kahteen<br />
ryhmään mediaanin kohdalta on lyhyemmän<br />
mutaation omaavien potilaiden eloonjäämisaika<br />
merkitsevästi pidempi kuin lyhyemmän<br />
mutaation omaavilla. (#3530) (kuva1).<br />
Näyttäisi siltä, että varsinaisen FlT3mutaatiolöydösen<br />
ohella olisi tarkoituksenmukaista<br />
sisällyttää laboratorioraporttiin<br />
tieto sekä mutatoituneen kloonin osuudesta<br />
näytteessä että duplikoituneen geenialueen<br />
pituudesta.<br />
AMl:n molekyyligeneettinen luokittelu<br />
tulee varmasti uudistumaan ja täydentymään<br />
aivan lähitulevaisuudessa. syväsekvenssointitekniikan<br />
käyttöönotto kliinisessä<br />
diagnostiikassa voi lähitulevaisuudessa<br />
mahdollistaa myös sen, että yksittäisten<br />
mutaatioiden analyysitarvetta ei tarvitse<br />
erikseen miettiä, vaan kaikki mahdollisesti<br />
relevantit mutaatiot voidaan pyytää laboratoriosta<br />
AMl-tutkimuspakettina. Mutta toistaiseksi<br />
vielä mutaatiotutkimukset pyydetään<br />
yksittäisinä tutkimuksina. Pitämässään koulutuksellisessa<br />
luennossa Prof. H. Döhner<br />
(Ulm) otti kantaa julkaistujen tutkimustulosten<br />
perusteella siihen, miltä osin tiettyjä<br />
mutaatiotutkimuksia voidaan jo pitää hyvään<br />
kliiniseen hoitokäytäntöön kuuluvina ja taas<br />
miltä osin mutaatioanalyysejä tulisi vielä<br />
tehdä vain kliinisen tutkimuksen puitteissa.<br />
Hyväennusteiseen AMl:aan liittyvien fuusiogeenien<br />
(PMl-rArA, rUNX1-rUNX1T1 ja<br />
CBFB-MyH11) suhteen negatiivisiksi jääviltä<br />
potilailta voidaan rutiiniluontoisesti suositella<br />
FlT3-pituusmutaation ja NPM1-mutaation,<br />
sekä ainakin edellisten jäädessä negatiivisiksi,<br />
CEBPA-geenin mutaatioiden tutkimista.<br />
Kaikkien näiden mutaatioiden tiedetään<br />
vaikuttavan oleellisesti potilaan prognoosiin<br />
ja jo tällä hetkellä on käytettävissä<br />
niin paljon tietoa, että näiden mutaatiolöydösten<br />
pitäisi vaikuttaa myös hoitovalintoihin.<br />
iDH1-, iDH2-, WT1-, rUNX1-, TET2- ja<br />
DNMT3A-mutaatiot ovat tulossa kliiniseen<br />
ASH 2011<br />
diagnostiikkaan, mutta vielä toistaiseksi ei<br />
ole olemassa riittävästi tutkimustuloksia siihen,<br />
että nämä mutaatiot (mahdollista jäännöstautianalyysinä<br />
käyttöä lukuunottamatta)<br />
vaikuttaisivat hoitovalintoihin. Mutaatioiden<br />
merkityksestä on olemassa eniten tietoa<br />
normaalikaryotyypin potilailla, sillä useat<br />
laajat tutkimukset ovat rajoittuneet nimenomaan<br />
normaalikaryotyypin AMl-potilaisiin.<br />
FlT3 ja NPM1-mutaatioilla on osoitettu<br />
olevan prognostista merkitystä myös poikkeavan<br />
karyotyypin potilailla, joten perusteita<br />
tutkimusten rajaamisella vain normaalin<br />
karyotyypin yhteyteen ei ole. AMl:n molekulaarinen<br />
diagnoosi nopeutuu huomattavasti,<br />
mikäli NPM1- ja FlT3-mutaatiotutkimukset<br />
voidaan tehdä diagnoosivaiheen näytteestä<br />
heti odottamatta karyotyypityksen valmistumista.<br />
Tämän seurauksena myös tulokset<br />
mahdollisista molekulaarisista jatkotutkimuksista<br />
(CEBPA, cKiT, NUP98-NsD1) saadaan<br />
varhaisemmassa vaiheessa ja mahdollisimman<br />
suuri osa potilaista saadaan molekulaarisen<br />
jäännöstautianalytiikan piiriin.<br />
All:n moleKYYligeneTiiKKA<br />
All:n kliiniseen käyttöön vakiintunut molekyyligenetiikka<br />
eroaa AMl:n molekyyligenetiikasta<br />
sikäli, että kattavasta molekulaarisesta<br />
jäännöstautianalytiikasta on All:ssa<br />
jo vuosikymmenen kokemus. All:ssa lähes<br />
kaikissa tapauksissa esiintyvien antigeenireseptorigeenien<br />
uudelleenjärjestymiin<br />
kohdentuva PCr-jäännöstautianalytiikka<br />
on tarjonnut mahdollisuuden kattavaan<br />
jäännöstautianalytiikkaan. Eurooppalaisen<br />
EuroMrD-konsortion kymmenen vuotta jatkuneen<br />
työn tuloksena klonaalisiin ig- tai<br />
TCr-geenien uudelleenjärjestymiin kohdistuva<br />
PCr-jäännöstautianalytiikka on erittäin<br />
hyvin standardoitua ja eurooppalaiset keskukset<br />
pystyvät tuottamaan eri tutkimusten<br />
välillä vertailukelpoista jäännöstautidataa.<br />
suomesta TyKslAB on ollut EuroMrD-konsortion<br />
jäsen viiden vuoden ajan. Molekulaarisen<br />
jäännöstautianalytiikan standardoinnissa<br />
eurooppalaiset laboratoriot ovat selkeästi<br />
edellä vastaavia amerikkalaisia laboratorioita,<br />
joilla edelleen jatkuva ongelma<br />
näyttää olevan, että jäännöstautianalytiikalla<br />
tuotetut tulokset eivät ole riittävän vertailukelpoisia,<br />
jotta niitä voitaisiin käyttää hoito-ohjelmien<br />
pohjana.
Kuva 1. KoKonaiseloonjääMinen ja TauTivapaa eloonjääMinen aMl-poTilailla,<br />
joilla FlT3-piTuusMuTaaTion dupliKoiTuneen alueen piTuus on<br />
(a)
30<br />
Eurooppalaisten All:n hoitokeskusten<br />
tulosten perusteella lähes kaikki perinteiset<br />
tunnetut All:n riskitekijät korreloivat niin<br />
voimakkaasti Aso-PCr:llä tietyissä hoidon<br />
alkuvaiheen aikapisteissä mitattuun jäännöstautiin,<br />
että aika näyttäisi olevan kypsä<br />
sille, että perinteisten riskimittareiden käytöstä<br />
voidaan luopua ja korvata ne molekulaarisen<br />
jäännöstaudin mittaamisella. Valitettavasti<br />
kuitenkaan edes molekulaarinen<br />
remissio ei ole täysin varma tae pysyvän<br />
remission saavuttamisesta, siksi tutkimustyö<br />
on kohdistunut voimakkaasti sellaisten<br />
ennustetekijöiden löytämiseen, jotka eivät<br />
korreloituisi molekulaariseen jäännöstautiin<br />
ja joiden avulla saataisiin jäännöstaudin<br />
määrästä riippumatonta ennakkotietoa<br />
relapsiriskistä. Useiden toisistaan riippumattomien<br />
tutkimusten perusteella näyttää<br />
siltä, että B-All:ssa ikaros-geenin (iKZF1)<br />
mikrodeleetiot liittyvät huonoon ennusteeseen<br />
(#409, #427, #566, #572, #741,<br />
#912, #1455, #3518). lisääntynyt relapsiriski<br />
ei ole kaikissa tapauksissa nähtävissä<br />
jäännöstaudin mittaamisella, sillä iKZF1mikrodeleetiopotilaat<br />
voivat hoidon alkuvaiheessa<br />
saada hyvän hoitovasteen, mutta<br />
myöhempi relapsiriski säilyy siitä huolimatta<br />
korkeana. T-All:n tunnetuista mutaatioista<br />
NoTCH/FBWX7-geenin mutaatiot liittyvät<br />
parempaan pitkän ajan hoitovasteeseen riippumatta<br />
siitä, vaikka hoidon alkuvaiheissa<br />
mitattu jäännöstauti Aso-PCr:llä mitattuna<br />
säilyisi korkealla tasolla. NoTCH/FBWX7geenin<br />
mutaatioita esiintyy yli puolella T-All<br />
-potilaista (#3551). Edellä olevan perusteella<br />
ranskalaisessa GrAAll-hoitotutkimuksessa<br />
on päädytty siihen, että kaikki Ph-negatiivisen<br />
All:n perinteiset riskiluokittelijat voidaan<br />
korvata KZF1/NoTCH/FBWX7-mutaatiotutkimuksilla<br />
sekä kolmen kuukauden<br />
kohdalla hoidon alusta tehtävällä herkällä<br />
EuroMrD-suositusten mukaisella Aso-PCr<br />
-jäännöstautitutkimuksella (#572). Tämän<br />
uuden riskiluokittelun perusteella noin puolet<br />
All-potilaista on sijoitettavissa hyväennusteiseen<br />
ryhmään, missä relapsiriski on<br />
tasoa 10–15 %.<br />
BCr-ABl1-fuusiogeenipositiivisuutta<br />
on pidetty merkittävänä huonoennusteisen<br />
All:n indikaattorina. Näillä Ph-positiivisilla<br />
All-potilailla iKZF1-mikrodeleetioita esiintyy<br />
suuremmalla frekvenssillä kuin Ph-nega-<br />
ASH 2011<br />
tiivisessa All:ssa. Kuitenkin myös Ph-positiivisilla<br />
All-potilailla iKZF1-deleetion avulla<br />
on mahdollista määritellä huonoennusteisempi<br />
alaryhmä. Aikuisiän All:ssa iKZF1mikrodeleetioita<br />
esiintyy lähes kolmanneksella<br />
potilaista, mutta pediatrisessa All:ssa<br />
esiintymisfrekvenssi jää alle 10 %:n tasolle<br />
liittyen ilmeisesti osaltaan Ph-positiivisen<br />
All:n harvinaisuuteen lapsilla. italialaisessa<br />
tutkimuksessa osoitettiin, että noin puolet<br />
pediatriseen Ph-negatiiviseen All:aan<br />
liittyvistä iKZF1-deleetioista on koko geenin<br />
deleetioita toisen puolen ollessa intrageenisiä<br />
mikrodeleetioita. Mielenkiintoinen löydös<br />
oli, että huono ennuste näytti liittyvän<br />
yksinomaan intrageenisiin mikrodeleetioihin.<br />
Mikäli koko iKZF1-geeni oli deletoitunut,<br />
relapsiriski oli kutakuinkin samalla tasolla<br />
kuin koko potilasaineistossa (#3518).<br />
Hollantilaisessa tutkimuksessa selvitettiin<br />
iKZF1-deleetioiden esiintyvyyttä Phpositiivisessa<br />
pediatrisessa All:ssa. 26<br />
potilaasta iKZF1-deleetio esiintyi 16:lla.<br />
iKZF1-posiitiivisilla potilailla relapsiriski oli<br />
lähes 60 %, kun se iKZF1-deleetionegatiivisilla<br />
potilailla oli 10 %. Perinteisistä riskitekijöistä<br />
iKZF1-deleetiopositiivisuus korreloi<br />
voimakkaasti diagnoosivaiheen veren leukosyyttitasoon<br />
(#3528).<br />
T-All -potilailla kokonaan oma diagnostinen<br />
ja vasta äskettäin määritelty alaryhmänsä<br />
näyttäisi olevan ETP- (early thymic<br />
progenitor) All, mikä immunofenotyypiltään<br />
on CD1a-, CD8-, CD5 heikko ja<br />
ilmentää lisäksi kantasolu- ja/tai myeloisia<br />
antigeenejä. T-solureseptorigeenit ovat yli<br />
puolessa tapauksista uudelleenjärjestäytymättömässä<br />
tilassa, minkä vuoksi yritykset<br />
Aso-PCr-jäännöstautianalyysin laatimiseksi<br />
voivat epäonnistua. NoTCH1-geeni on<br />
mutatoitunut vain noin 10–20 %:ssa tapauksista,<br />
mutta FlT3-geenissä on pituusmutaatio<br />
noin kolmanneksella ETP-All -potilaista,<br />
kun muuten FlT3-mutaatio on hyvin<br />
harvinainen (n. 1 %) T-All:ssa. Potilaiden<br />
ennuste on hyvin huono, mikäli hoito toteutetaan<br />
tavanomaisen T-All -hoidon mukaisesti<br />
(#3551).<br />
Käytettäessä jäännöstautianalyysejä<br />
hoidon myöhäisemmässä vaiheessa ig-<br />
ja TCr-uudelleenjärjestymiin kohdistuvat<br />
Aso-PCr -tutkimukset eivät ole täysin luotettavia<br />
kehittyvää relapsia poissulkevia tut-
kimuksia, sillä relapsikloonista em. markkerit<br />
voivat puuttua. Tilannetta voidaan korjata<br />
käyttämällä PCr-kohteena samanaikaisesti<br />
kahta tai useampaa diagnoosivaiheessa<br />
tunnistettua uudelleenjärjestymää, jolloin<br />
todennäköisyys väärälle negatiiviselle<br />
löydökselle pienenee.<br />
Tyypillistä All:lle on, että samaankin<br />
geeniin kohdistuvia uudelleenjärjestymiä<br />
voi diagnoosivaiheessa löytyä useita. Konsensus-PCr:n<br />
avulla klooneja voidaan yleensä<br />
tunnistaa noin 1–5 %:n herkkyystasolle<br />
saakka, mutta syväsekvenssoissa detektioraja<br />
alenee käytännössä lähes samalle tasolle<br />
saakka kuin mikä on ollut käytetty sekvenssointisyvyys.<br />
syväsekvenssointi onkin<br />
paljastanut, että All:n diagnoosivaiheessa<br />
erilaisia subklooneja voi löytyä satoja, ja<br />
myös näiden subkloonien immunofenotyypit<br />
voivat erota toisistaan. Näin on ymmärrettävissä,<br />
että jonkin yksittäisen uudelleenjärjestymän<br />
tai immunofenotyypin käyttö syöpäsolun<br />
tunnisteena ei riitä tilanteeseen,<br />
jossa relapsi kehittyykin jostain muusta kloo-<br />
ASH 2011<br />
31<br />
nista, kuin diagnoosivaiheen valtakloonista.<br />
Mikäli subkloonin määritelmänä käytetään<br />
sitä, että sama uudelleenjärjestymä on tunnistettavissa<br />
vähintään 5 %:sta soluja, vähenee<br />
subkloonien määrä oleellisesti ja vastaa<br />
sitä tietämystä, mikä aiemmin on saatu konsensus-PCr:ää<br />
käyttämällä (#1436).<br />
Aso-PCr:ään verrattuna syväsekvenssointi<br />
tarjoaa sen edun, että jäännöstaudin<br />
seurannan ei tarvitse rajoittua vain yhteen<br />
tai muutamaan subklooniin, vaan periaatteessa<br />
kaikkia, vähäisiäkin diagnoosivaiheen<br />
klooneja voidaan sisällyttää jäännöstautiseurantaan.<br />
Myös mahdollisten uusien<br />
kehittyvien kloonien diagnostiikka tulee<br />
mahdolliseksi ja alun perin vähäisen subkloonin<br />
aiheuttama relapsi ei tule yllätyksenä.<br />
Useiden subkloonien aiheuttamat<br />
ongelmat Aso-PCr -jäännöstautianalytiikalle<br />
näyttäisivät koskevan vain akuuttia lymfoblastileukemiaa.<br />
Muissa lymfoproliferatiivisissa<br />
taudeissa (lymfoomat, Kll, multippeli<br />
myelooma) diagnoosivaiheen uudelleenjärjestymä<br />
on varsin stabiili ja siihen kohdistuva<br />
Kuva 2. TauTiKloonin MääriTelMä: >5% KaiKisTa soluisTa ja vähinTään 1000 MoleKyyliä<br />
igh-geenin suhteen uudelleenjärjestyneiden kloonien lukumäärä akuutissa lymfoblastileukemiassa. (#1436).<br />
NäyTTEiDEN lUKUMäärä<br />
20<br />
15<br />
10<br />
5<br />
0<br />
1 2 3 4 5<br />
KlooNiEN lUKUMäärä / NäyTE
32<br />
Aso-PCr soveltuu hyvin käytettäväksi myös<br />
relapsia ennakoivana tutkimuksena (#1436)<br />
(kuva 2).<br />
mulTiPPelin mYeloomAn<br />
moleKulAARinen<br />
JäännöSTAuTiAnAlYTiiKKA<br />
immunoglobuliinigeenin raskaan ketjun klonaaliseen<br />
uudelleenjärjestymään kohdentuvaa<br />
Aso-PCr -jäänöstautianalyysiä käytetään<br />
maailmalla edelleen suhteellisen<br />
harvoin multippelin myelooman yhteydessä,<br />
vaikka suomessa analytiikalla on pitkä<br />
perinne. TyKs:n laboratoriossa kvantitatiivisen<br />
Aso-PCr:n kliininen soveltaminen<br />
alkoi 15 vuotta sitten nimenomaan tarpeesta<br />
jäännöstaudin mittaamiseen autologisen<br />
siirron saaneilla myeloomapotilailla. Myös<br />
uusimmat kansalliset myeloomatutkimuksemme<br />
soveltavat Aso-PCr:ää parhaimman<br />
hoitovasteen saaneilla potilailla.<br />
italialaisessa tutkimuksessa sovellettiin<br />
EuroMrD-suositusten mukaista Aso-PCr:ää<br />
autologisen siirron ja bortetsomibi/talido-<br />
ASH 2011<br />
midi/deksametasoni konsolidaatiohoidon<br />
saaneilla myeloomapotilailla. Alle 0,01 %:n<br />
jäännöstautitason saavuttaneilla potilailla<br />
ensimmäiset tautiprogressiot todettiin vasta<br />
viiden vuoden seurannan jälkeen, kun heikomman<br />
molekulaarisen vasteen saaneista<br />
potilaista tauti oli progredioitunut vastaavan<br />
ajan kuluttua jo noin puolella (#827). saksalaisessa<br />
tutkimuksessa EuroMrD-kriteerein<br />
standardoitua Aso-PCr:ää käytettiin monitorointianalyysinä<br />
73:lle myeloomapotilaalle,<br />
jotka olivat saaneet autologisen-allogeenisen<br />
tandemsiirron. Tulosten perusteella<br />
tällaisessa hoitostrategiassa selkeäksi hoitotavoitteeksi<br />
on otettava molekulaarisen<br />
remission saavuttaminen. Vähäinenkin jäännöstauti<br />
analyysin herkkyysrajan tuntumassa<br />
(MrD mixed, vaihtelua eri seurantanäytteissä<br />
positiivisen ja negatiivisen välillä) riitti<br />
pudottamaan progressiovapaan eloonjäämisen<br />
lähelle MrD-positiivista ryhmää (#148)<br />
(kuva3).<br />
japanilaisessa tutkimuksessa Aso-<br />
PCr -analytiikkaa oli sovellettu myelooma-<br />
Kuva 3. auTologisella-allogeenisella TandeMsiirrolla hoideTTujen<br />
MyelooMapoTilaiden progressiovapaa eloonjääMinen (n=73) (#148)<br />
ETENEMisVAPAA ElossAolo<br />
1,0<br />
0,8<br />
0,6<br />
0,4<br />
0,2<br />
0,0<br />
p
potilaille siten, että klonaalinen uudelleenjärjestymä<br />
oli analysoitu muun diagnoosivaiheen<br />
materiaalin puutteessa aluslaseille<br />
tehdystä sivelyvalmisteesta raaputetusta<br />
näytemateriaalista (#1879). Klonaalinen<br />
uudelleenjärjestymä oli tunnistettavissa<br />
ja sekvenssoitavissa suurimmassa osassa<br />
tapauksista myös tällaisesta materiaalista,<br />
mutta EuroMrD-suositusten mukainen<br />
standardoitu jäännöstautianalytiikka ei ole<br />
näin mahdollista, sillä materiaalia ei saada<br />
riittävästi kvanti<strong>fi</strong>oinnin perustana olevien<br />
laimennussarjojen valmistamiseen. Mahdollinen<br />
tuleva diagnoosivaiheen materiaalin<br />
tarve tulisi siksi muistaa ennen hoitojen<br />
aloittamista. luuytimen mononukleaariset<br />
solut voidaan suhteellisen vähäisin kustannuksin<br />
eristää diagnoosivaiheen näytteestä<br />
ja säilyttää laboratoriossa mahdollisia tulevia<br />
analyysitarpeita varten.<br />
moleKYYligeneeTTiSTen<br />
TuTKimuSTen KäYTTö RelAPSin<br />
ennAKoimiSeen AllogeeniSen<br />
SiiRRon JälKeen<br />
ig- ja TCr-uudelleenjärjestymiin All:ssa<br />
sekä FlT3-mutaatioon AMl:ssa liittyviä<br />
rajoituksia lukuun ottamatta edellä käsitellyt<br />
jäännöstautitutkimukset sopivat käytettäväksi<br />
myös relapsia ennakoivina tutkimuksina<br />
allogeenisen kantasolusiirron jälkeen.<br />
Näiden lisäksi samaan tarkoitukseen on käytetty<br />
PCr-kimeratutkimusta ja WT1-geenin<br />
ekspressiotason määrittämistä joko veri-<br />
tai luuydinnäytteestä. relapsi voi kehittyä<br />
nopeasti, joten ennakoivaa tutkimusta tulisi<br />
kriittisessä hoidon vaiheessa toistaa tihein<br />
väliajoin. Tämän vuoksi ihanteellisimmassa<br />
tapauksessa tutkimuksen tulisi olla tehtävissä<br />
verinäytteestä. Verestä ja luuydinnäytteestä<br />
saavutettavissa analyysin herkkyysta-<br />
ASH 2011<br />
33<br />
soissa on kuitenkin tautikohtaisia eroja. Esimerkiksi<br />
B-All:ssa luuydinnäytteestä saavutetaan<br />
keskimäärin 14 kertaa parempi herkkyys<br />
kuin verinäytteestä (#2542), kun taas<br />
T-All:ssa, KMl:ssa ja APl:ssa verestä on<br />
saavutettavissa kutakuinkin sama jäännöstautianalyysin<br />
herkkyystaso kuin luuytimestä.<br />
Kimera-analyysi tehdään yleensä verinäytteestä<br />
ja fragmenttianalyysinä toteutettuna<br />
tutkimuksen herkkyys jää noin 1 %:n<br />
tasolle. Näin toteutettuna kimera-analyysin<br />
käyttöarvo relapsia ennakoivana tutkimuksena<br />
ei ole kovin hyvä. Kimera-analyysin käyttökelpoisuus<br />
paranee, mikäli analyysi tehdään<br />
CD34+ solufraktiosta. riittävän ja tarpeeksi<br />
puhtaan CD34+ solumäärän erottamiseksi<br />
analyysi on tehtävä luuydinnäytteestä,<br />
mikäli käytettävissä ei ole soluja erottelevaa<br />
virtaussytometriä.<br />
WT1-ekspressioanalyysin on aiemmin<br />
osoitettu olevan käyttökelpoinen relapsia<br />
ennakoiva tutkimus, ja sen käyttökelpoisuus<br />
osoitettiin myös kahdessa AsH 2011<br />
-kongressissa julkaistussa tutkimuksessa<br />
(#2507, #4102). WT1-ekspressioanalyysin<br />
kliininen käyttöarvo näyttäisi olevan selvästi<br />
parempi, kuin mitä olisi ennakoitavissa WT1analyysin<br />
suhteellisen vaatimattomasta noin<br />
1 %:n herkkyydestä jäännöstautianalyysinä.<br />
Epäsuorana johtopäätöksenä olisi, että<br />
relapsin alkuvaiheessa tautisolujen WT1-ekspressio<br />
olisi erittäin korkealla tasolla. WT1ekspressioanalyysi<br />
on nykyisin jo hyvin standardoitu<br />
ja analyysin teknistä suorittamista<br />
varten on olemassa European leukemia<br />
Netin julkaisema konsensussuositus, joten<br />
WT1-ekspressiotutkimuksen käyttömahdollisuus<br />
ansaitsisi huomiota ainakin niillä<br />
allogeenisen siirron saaneilla potilailla, joilla<br />
ei ole käytettävissä mitään muuta herkkää<br />
molekulaarista jäännöstaudin markkeria.
mika Kontro, HYKS:<br />
AMl:N KoHDENNETUT HoiDoT TUlossA
AMl:n osalta AsH-kokouksen keskeisiä teemoja olivat genetiikka ja leukeemiset<br />
kantasolut. Uusia, kehittelyssä olevia kohdennettuja hoitoja käsiteltiin<br />
myös laajasti.<br />
Ensimmäinen AMl-genomi sekvensoitiin<br />
vuonna 2008. Työ vei kaksi vuotta ja rahaa<br />
kului 1,6 miljoonaa dollaria. sittemmin kustannukset<br />
ovat pudonneet ja vauhti on kiihtynyt<br />
– nyt AMl-genomin sekvensointi on tehty<br />
jo lähes sadalle potilaalle, ja suppeampia<br />
eksomisekvensointeja on tehty moninkertainen<br />
määrä. Vaikka uusia mutaatioita on<br />
löydetty runsaasti, on AMl paljastunut luultua<br />
yksinkertaisemmaksi taudiksi; potilaalla<br />
todettavien mutaatioiden määrä on keskimäärin<br />
vain 8–10. Tiuhojen julkaisujen ”The<br />
gene of the month” -aika alkaa hiljalleen<br />
olla ohi, ja seuraavina vuosina keskitytään<br />
mutaatioiden funktionaaliseen ja ennusteelliseen<br />
merkitykseen. Hoidollisia sovelluksia<br />
on odotettu pitkään ja niitä käsiteltiinkin<br />
kokouksessa laajasti.<br />
FlT3-inHibiiTToRiT – uuSi SuKuPolvi<br />
laajakirjoiset, myös FlT3-proteiinia estävät<br />
tyrosiinikinaasiestäjät ovat toimineet<br />
tienraivaajina AMl:n kohdennetuissa hoidoissa.<br />
Tulokset eivät kuitenkaan ole olleet<br />
odotusten mukaisia; sorafenibin menestys<br />
on rajoittunut anekdotaalisiin tapausselostuksiin,<br />
eikä lestaurtinibi puolestaan onnistunut<br />
osoittamaan elinaikahyötyä vuosi sitten<br />
julkaistussa randomoidussa tutkimuksessa<br />
(Blood 2011;117: 3294–3301). on jo<br />
pohdittu, onko FlT3-iTD -mutaatiolla edes<br />
merkitystä AMl:n patogeneesissä vai onko<br />
kyseessä vain ns. passenger-mutaatio.<br />
AC220 (kvitsartinibi) on lupaava toisen<br />
polven oraalinen FlT3/KiT-inhibiittori. Posterissa<br />
(#2576) esitettiin alustavia tuloksia<br />
kansainvälisestä faasi ii -monikeskustutkimuksesta.<br />
Tutkimuksessa oli mukana sekä<br />
yli 60-vuotiaita relapsoituneita/refraktaareja<br />
potilaita (kohortti 1) sekä nuorempia (yli<br />
18-vuotiaita) potilaita, jotka olivat refraktaareja/relapsoituneita<br />
toisenkin linjan hoidol-<br />
ASH AsH 2011<br />
35<br />
le (kohortti 2). Potilaat saivat tätä oraalista,<br />
hyvin siedettyä lääkettä monoterapiana.<br />
Vastetta (Crc = Cr+Crp+Cri) saavutettiin<br />
haastavassa potilasaineistossa 45 %:lla. refraktaareista<br />
potilaista peräti 62 % sai Crctasoisen<br />
vasteen hoidolle. Kaikkien potilaiden<br />
elossaoloajan mediaani oli 24,7 viikkoa.<br />
Mutta onko FlT3 hoidon kohde vai vain<br />
mukana ollut matkustaja ja markkeri? Kalifornialaiset<br />
tutkijat poimivat käynnissä olevasta<br />
AC220-tutkimuksesta FlT3-positiivisia<br />
potilaita, jotka saavuttivat AC220-hoidolla<br />
remission ja sittemmin relapsoituivat.<br />
Näiden yhdeksän potilaan parinäytteistä<br />
(näytteet ennen hoitoa ja relapsissa) etsittiin<br />
muutoksia FlT3-reseptorista. Kaikista<br />
relapsivaiheen näytteistä löydettiin AC220resistenssin<br />
aiheuttavia mutaatioita. Tutkimus<br />
osoittaa FlT3-iTD:n olevan käypä hoidon<br />
kohde ja sen, että AC220:n vaikutus<br />
perustuu FlT3:n estoon (#937).<br />
mll-FuuSiogeeniT - KoHdenneTTuJA<br />
HoiToJA KeHiTeTään<br />
Mll-geenin (mixed lineage leukemia) translokaatiot<br />
johtavat aggressiivisiin akuutteihin<br />
leukemioihin, joilla on huono pitkäaikaisennuste.<br />
Vaikka Mll-geeni voi fuusioitua yli<br />
50 geenin kanssa, valtaosassa fuusioita on<br />
mukana transkriptiotekijä sEC (super elongation<br />
complex). Havainto on vienyt kohdennettujen<br />
hoitojen kehitystä nopeasti eteenpäin.<br />
Kokouksessa esitettiin lukuisia kohdennettuja<br />
hoitoja Mll-geenifuusion aiheuttamaan<br />
leukemiaan (#55, #58, #2497,<br />
#2500). Ehkä mielenkiintoisimpana esiteltiin<br />
uusi pienimolekylaarinen BET-inhibiittori<br />
(i-BET-151, GsK), jolla estetään fuusiosta<br />
aiheutuvaa poikkeavaa transkriptiota. BETinhibiittorilla<br />
saavutettiin vastetta sekä in vitro<br />
-kokeissa että Mll-leukemian (Mll-AF4 ja<br />
Mll-AF9) hiirimallissa (#55). Usealla saral-
36<br />
la etenevät tutkimukset johtanevat lähivuosina<br />
uusien molekyylien kliinisiin tutkimuksiin.<br />
dnmT3A-muTAATioT –vASTeTTA<br />
HYPomeTYloiville lääKKeille?<br />
DNMT3A-mutaation yleisyys ja ennusteellinen<br />
merkitys AMl:ssa kuvattiin vuonna 2010<br />
(NEjM 2010;363:2424–2433). Mutaatioita<br />
todettiin kaikkiaan 22 %:lla potilaista, ja ne<br />
olivat keskittyneet lähes yksinomaan keskiriskin<br />
tauteihin. DNMT3-mutaatiot assosioituvat<br />
FlT3-, NPM1- ja iDH 1/2 -mutaatioihin,<br />
ja DNMT3A-mutaatioihin todettiin liittyvän<br />
ennustetta huonontava vaikutus. Vastaavankaltaisia<br />
tietoja on julkaistu edeltävän<br />
vuoden aikana (jCo 2011; 29:2889–2896).<br />
Kokouksessa AMlsG-ryhmä syvensi<br />
tietoa mutaation ennusteellisesta merkityksestä.<br />
Tutkijat kävivät läpi 1218 potilaan<br />
aineiston (ikä 18–61 vuotta, seurantaajan<br />
mediaani 5,06 vuotta), joista mutaatio<br />
todettiin 19,6 %:lla. DNMT3A-mutaatioita<br />
todettiin varsinkin iäkkäämmillä potilailla<br />
ja naisilla. Vaikka DNMT3A-mutaatio ei vaikuttanut<br />
Cr:n saavuttamiseen, EFs:aan tai<br />
rFs:aan todettiin kuitenkin ennustetta huonontava<br />
vaikutus sytogenenetiikaltaan normaalissa<br />
taudissa (CN-AMl). Monimuuttuja-analyysissä<br />
todettiin ennusteen huonontuvan<br />
nimenomaan ”epäsuotuisamman”<br />
molekyyligenetiikan taudeissa (NPM1-wt<br />
riippumatta FlT3-iTD-statuksesta, NPM1mutaatio<br />
ja FlT3-iTD, CEBPA-wt) (#415).<br />
Myös mutaation patofysiologisesta merkityksestä<br />
saatiin lisätietoa: DNMT3A-mutaatio<br />
johtaa hematopoieettisten kantasolujen<br />
erilaistumisen estoon (#386).<br />
Mutaation yleisyyden ja ennustevaikutuksen<br />
vuoksi hoidollisia sovellutuksia<br />
on odotettu. Koska DNMT3A on keskeinen<br />
DNA:n metylaation säätelijä, on arveltu, että<br />
hypometyloivilla hoidoilla saavutetaan hoitovastetta.<br />
ohiolaisten tutkijoiden pieni aineisto<br />
oli tässä suhteessa lupaava. Desitabiinihoidolla<br />
saavutettiin iäkkäiden potilaiden<br />
AMl:ssa Cr-vaste kuudella potilaalla kahdeksasta,<br />
joilla oli DNMT3A-mutaatio (13/39<br />
DNMT3A-wt ryhmässä; p=0,05). Kaikki vii-<br />
ASH 2011<br />
si potilasta, joilla todettiin NPM1- ja FlT3mutaatiot<br />
saavuttivat Cr-vasteen (p=0,008).<br />
Myös elinaikaetu todettiin (16,8 vs. 11,0 kk),<br />
mutta pienen potilasmäärän vuoksi se ei<br />
ollut tilastollisesti merkitsevä (#944).<br />
vARHAiSeT Aml-KAnTASoluT<br />
Molekyyligenetiikan huomattavista edistysaskelista<br />
huolimatta on edelleen ollut epäselvää,<br />
mitkä muutokset johtavat leukemian<br />
syntyyn. Plenary-sessiossa esitetyssä<br />
abstraktissa stanfordilaiset tutkijat valottivat<br />
leukemogeneesin mysteeriä (#4). AMlpotilaiden<br />
leukeemisen solukon mutaatiot<br />
tutkittiin eksomisekvensoinnilla. löydettyjä<br />
mutaatioita verrattiin samojen potilaiden<br />
”terveissä” hematopoieettisissa kantasoluissa<br />
todettuihin. Valtaosa mutaatiosta,<br />
muun muassa FlT3 iTD - ja iDH1 r132H<br />
-mutaatiot, olivat todettavissa ainoastaan<br />
leukeemisissa soluissa. Tietyt mutaatiot<br />
olivat kuitenkin todettavissa myös osassa<br />
hematopoieettisia kantasoluja. Tällaisia<br />
“varhaisia” mutaatioita olivat muun muassa<br />
NPM1c- ja TET2-mutaatiot sekä tietyt<br />
mutaatiot solusykliä säätelevissä geeneissä.<br />
Tutkijat selvittivät edelleen löydettyjen<br />
preleukeemisten kloonien määrää yksittäisten<br />
solujen sNP-analyysien avulla. Potilaskohtaiset<br />
erot olivat suuria, mutta tavallisten<br />
hematopoieettisten kantasolujen määrä<br />
oli 6–50-kertainen preleukeemiseen klooniin<br />
verrattuna.<br />
Elegantti tutkimusasetelma paljastaa<br />
siis preleukeemisissa kantasoluissa tapahtuvien<br />
uusien, ”myöhäisten” mutaatioiden<br />
johtavan varsinaiseen leukemiaan. Tutkimustulokset<br />
valottavat myös relapsin syitä –<br />
sen lisäksi, että leukeeminen jäännössolukko<br />
voi aiheuttaa relapsin, saattaa relapsin<br />
taustalla olla myös preleukeemisen kloonin<br />
evoluutio leukeemiseksi. Tällöin on todennäköistä,<br />
että relapsoituneen taudin genotyyppi<br />
poikkeaa diagnoosin yhteydessä todetusta<br />
(kuva 1). Tutkijat arvelevat, että preleukeemiseen<br />
klooniin kohdennettu hoito<br />
tarjoaisi uusia mahdollisuuksia leukemian<br />
ennusteen parantamiseen.
Kuva 1. esiTeTTy Malli leuKeMian relapsiin johTavasTa KlonaalisesTa evoluuTiosTa<br />
DiAGNoosi<br />
lEUKEMiA<br />
HEMAToPoiEETTisET<br />
KANTAsolUT<br />
HoiTo<br />
C<br />
B<br />
A<br />
D<br />
ranskalainen alFa-ryhmä liitti gemtutsumabi-otsogamiinin (go) 50–70-vuotiaiden de novo-aMl -potilaiden tavanomaiseen<br />
(araC+dnr) hoitoon. go annosteltiin induktiossa fraktioituna päivinä 1, 4 ja 7 annoksella 3mg/m 2 /pvä. Konsolidaatiohoidoissa<br />
potilaat saivat go:n päivänä 1 (3mg/m 2 ). Kahden vuoden kohdalla eFs parani (16,5 % vs. 41,4 %, p=0,0018).<br />
Myös elinaikaennuste parani hieman (os 2 vuotta, 43,5 % vs. 53,1 %). ennusteen paraneminen todettiin vain pienen ja<br />
keskiriskin taudeissa. hoitokuolleisuus ei lisääntynyt, mutta go-ryhmässä esiintyi enemmän pitkittyvää trombopeniaa (n.<br />
7 %:lla potilaista) ja vod:ia (n. 1 %:lla potilaista) (#6).<br />
pohjoisamerikkalaistutkijat esittivät tuloksia apl:n ylläpitohoidosta. hoito-ohjelmassa verrattiin kahta ylläpitohoitoa: aTraa<br />
monoterapiana (45 mg/m 2 7 päivän jaksoissa vuoroviikoin) tai aTraa yhdistettynä solusalpaajiin (6-Mp, 60 mg/<br />
m 2 ,päivittäisenä ja MTX ,20 mg/m 2 , viikoittaisena). ylläpitohoitoa jatkettiin vuoden ajan. MTX-Mp:n lisääminen apl:n ylläpitohoitoon<br />
ei parantanut elinaikaennustetta (p=0,33), mutta lisäsi tauditonta elinaikaa (#258).<br />
ASH 2011<br />
rElAPsi<br />
leukeemiset<br />
kloonit<br />
preleukeemiset<br />
kantasolukloonit<br />
37<br />
normaalit<br />
hematopoieettiset<br />
kantasolut<br />
A vallitseva leukeeminen klooni<br />
B Määrältään vähäinen leukeeminen<br />
klooni<br />
C leukeeminen klooni, lisämuutoksia<br />
perimässä<br />
D preukeeminen klooni, lisämuutoksia<br />
perimässä
Tero Pirttinen, TaYS:<br />
MYELODYSPLASTISET OIREYHTYMÄT – UUTTA TIETOA<br />
PERIMÄN MUUTOKSISTA JA HOITOVAIHTOEHDOISTA
Myelodysplastisiin oireyhtymiin (MDs) liittyvien geneettisten muutosten<br />
kirjo ja niiden merkitys täsmentyy palanen kerrallaan. Mutaatioita löytyy<br />
erityisesti epigeneettisen säätelyn alueelta (DNA:n ja histonien metylaatiota<br />
säätelevien entsyymien geeneistä) sekä mrNA:n prosessointiin liittyvistä<br />
proteiineista. Perimän muutosten tarkka tunteminen on edellytyksenä<br />
täsmähoitojen kehittämiselle.<br />
TieTo mdS:n PeRimän muuToKSiSTA<br />
TARKenTuu<br />
Edistyneellä sekvensoinnilla ehjän näköisistä<br />
kromosomeista löytyy mikrodeleetioita ja<br />
translokaatioita, joita edeltänee kromotripsis,<br />
kromosomin pirstoutuminen ja enemmän tai<br />
vähemmän hajamielinen uudelleenkokoaminen.<br />
sama patomekanismi saattaa aiheuttaa<br />
uniparentaalista disomiaa, missä mutatoituneen<br />
geenin alleelit ovat toistensa kopioita eli<br />
”samalta vanhemmalta” (UPD; kopiomäärältään<br />
neutraali heterotsygoottisuuden puutos;<br />
vertaa esim. CEBPA/AMl ja jAK2/PV).<br />
yleisin de novo MDs:n sytogeneettinen<br />
muutos on deleetio 5q. Distaalinen deleetio<br />
tuottaa tavallisen hyväennusteisen Del5qfenotyypin,<br />
proksimaaliseen deleetioon taas<br />
liittyy korkea transformaatioriski. Tautisoluun<br />
jääneestä 5q-alueesta ei sekvensoimalla näytä<br />
löytyvän mutaatioita, ja eläinkokeiden perusteella<br />
taudin fenotyyppi näyttäisikin selittyvän<br />
useiden geenien samanaikaisella haploinsuf<strong>fi</strong>sienssilla<br />
(geenin alentunut ekspressio toisen<br />
alleelin puuttuessa). ribosomaalisen proteiinin<br />
(rPs14) puutos aiheuttaa punasolulinjan<br />
häiriön, mir-145:n ja mir-146:n puutos trombosytoosia,<br />
EGr1:n puutos kantasolujen kiihtynyttä<br />
uusiutumista, ja HsPA9:n puutos erytrooisen<br />
kypsymisen hidastumista ja apoptoosin<br />
lisääntymistä. sPArC-geenin haploinsuf<strong>fi</strong>sienssi<br />
kiinnittää Del5q-kantasolut tiukemmin<br />
mikroympäristöönsä, mikä muodostaa<br />
kilpailuedun normaaleja kantasoluja vastaan.<br />
Ainakin osa lenalidomidin tehosta perustuu<br />
sen kykyyn lisätä sPArC-ilmentymää (#790).<br />
lenalidomidi pidentää elinaikaa transfuusioriippuvaisilla<br />
Del5q-potilailla lisäämättä<br />
transformaation riskiä (#119).<br />
ASH AsH 2011<br />
39<br />
Kromatiinin rakenne vaikuttaa geenien<br />
ilmentymiseen. MDs-solujen DNA:n<br />
metylaatiopro<strong>fi</strong>ili poikkeaa normaalista ja<br />
hypermetyloitujen alueiden transkriptioaktiivisuus<br />
on matala. Tärkein DNA:n sytosiini-emäksiä<br />
metyloiva entsyymi on DNAmetyylitransferaasi-3A<br />
(DNMT3A). Tunnetut<br />
mutaatiot aiheuttavat entsyymiaktiivisuuden<br />
pienentymisen ja liittyvät huonompaan<br />
ennusteeseen. TET-entsyymit hydroksyloivat<br />
metyloidun sytosiinin (5mC -> 5hmC),<br />
ja reaktioon tarvittavaa alfa-ketoglutaraattia<br />
tuottaa iDH-entsyymi. iDH-mutaatioiden<br />
kantajien ennuste on osoitettu keskimääräistä<br />
huonommaksi. AMl-potilailla iDH-mutaatiot<br />
ja TET2-mutaatiot ovat toisensa poissulkevia,<br />
ja molempien vaikutus DNA:n metylaatiopro<strong>fi</strong>iliin<br />
on sama. Miksi sitten ”hypometyloivien”<br />
lääkehoitojen vaikutus potilaiden<br />
ennusteeseen on myönteinen, jos ”hypometyloivien”<br />
mutaatioiden vaikutus on kielteinen?<br />
Vaikkakin MDs-potilaiden metylaatiopro<strong>fi</strong>ili<br />
poikkeaa normaalista, ei tautisolujen<br />
DNA ole yleisesti sen enemmän metyloitunutta<br />
kuin muidenkaan. Hoito atsasitidiinilla<br />
(AZA) ja desitabiinilla ei myöskään näytä<br />
alentavan DNA:n kokonaismetylaatioastetta,<br />
joten niiden nimittäminen hypometyloiviksi<br />
lääkkeiksi on osin harhaanjohtavaa.<br />
Myös histoniproteiinien metylaation ja<br />
asetylaation aste vaikuttaa kromatiinin rakenteeseen<br />
ja siten geeniekspressioon. EZH2entsyymi<br />
metyloi histoneja H3 ja H1 aiheuttaen<br />
transkriptioaktiivisuuden alentumista,<br />
ja MDs-potilailta löytyviin EZH2-mutaatioihin<br />
liittyy tavallisesti entsyymiaktiivisuuden<br />
lasku sekä alentunut elinajan ennuste. Muita<br />
paikalliseen transkriptioaktiivisuuteen vai-
40<br />
kuttavia entsyymejä ovat AsXl1, demetylaasi<br />
UTX ja muut jmjC-domainin sisältävät entsyymit,<br />
joiden geeneistä MDs-potilailla löytyy<br />
mutaatioita. Monilinjaisessa dysplasiassa on<br />
tärkeän transkriptiofaktorin PU.1:n ekspressio<br />
alentunut, ja tämä näyttäisi aiheutuvan<br />
histonimetylaatiosta; kokeellisesti metylaatioinhibiittorilla<br />
on saatu korjattua PU.1- ekspressiota<br />
ja edistettyä granulosyyttien differentiaatiota<br />
(#2783).<br />
spliseosomit ovat tuman rakenteita, jotka<br />
pilkkovat pre-mrNA:sta intronien osuudet<br />
pois tuottaen varsinaista lähetti-rNA:ta.<br />
spliseosomien U2 ja U12 rakenneproteiinilla<br />
sF3B1:lla näyttää olevan merkittävä rooli<br />
rengassideroblastisten MDs:ien (rArs,<br />
rCMD-rs) patogeneesissä (#3, #457; myös<br />
Blood 2011 118: 6239–6246 sekä N Engl j<br />
Med 2011; 365: 1384–1395). Malcovatin ja<br />
Papaemmanuilin ryhmä tutki MDs-potilailta<br />
sF3B1-geenin mutaatioiden ja mm. rengassideroblastien<br />
esiintyvyyttä. 150 potilasta<br />
533:sta (28,1 %) kantoi geenin somaattista<br />
mutaatiota, ja mutaatioiden kantajuus oli selvästi<br />
yleisempää rengassideroblastisten tautien<br />
kuin muiden WHo-luokituksen tautien<br />
kantajilla (72,0 % vs. 9,8 %, p
Kuva 1. epigeneeTTisTen reiTTien geneeTTiseT MuuToKseT MyelodysplasTisTa<br />
oireyhTyMää (Mds) sairasTavilla<br />
AsXl1<br />
EZH2<br />
HisToNi<br />
H3<br />
K27<br />
me3<br />
UTX<br />
jmjC<br />
ASH 2011<br />
DNMT3A<br />
isositraatti<br />
iDH1/2<br />
aKG<br />
5mC 5hmC ?<br />
TET2<br />
Kuvassa esitetään tiettyjen histonimodi<strong>fi</strong>kaation ja dna:n metylaation kannalta keskeisten tekijöiden normaali toiminta.<br />
Vasen reuna: näiden reittien mutaatioiden biologisia seurauksia ei useimmissa tapauksissa tunneta täysin. nukleosomeissa<br />
esiintyvän histoni h3:n karboksiterminaalisen pään lysiini-27:n (K27) trimetylaatio (me3) on yhteydessä transkription<br />
estymiseen. asXl1 on prC-ryhmän (polycomb repressive complex) proteiini, joka ylläpitää repressiota. Mdspotilailla<br />
on usein mutaatioita asXl1- ja eZh2-geeneissä, jotka koodaavat h3K27-metyylitransferaasia. Mds-potilaiden<br />
näytteissä on myös todettu harvoin mutaatioita (deleetioita) h3K27-demetylaasientsyymeissä, mm. uTX-entsyymissä ja<br />
muissa jmjC-domeenin sisältävissä proteiineissa.<br />
Oikea reuna: sytosiinin metylaatio Cpg-saarekkeissa on yhteydessä transkription estymiseen. dnMT3a on de novo<br />
dna-metyylitransferaasi, joka muuntaa metyloitumatonta sytosiinia 5mC:ksi. 5mC muuntuu TeT-proteiinien vaikutuksesta<br />
5hmC:ksi, kun idh-entsyymien tuottamaa aKg:ta on saatavilla. Mds-potilaiden näytteissä todetaan usein dnMT3a-,<br />
TeT2-, idh1- ja idh2-geenien mutaatioita.<br />
41
ulla Pihkala, HYKS:<br />
VirUsTorjUNTAoHjElMA<br />
KANTAsolUjENsiirToPoTilAillE
ekisteriluovuttajalta saadun kantasolujensiirron jälkeen virusinfektiot<br />
muodostavat merkittävän ja yleisen ongelman (39 %), joka on esiintymiseltään<br />
samaa suuruusluokkaa kuin malignin perustaudin relapsi (39 %).<br />
Keuhkotoksisuutta ilmenee 15 %:lla, ja bakteerisepsis 7 %:lla (Biol Blood<br />
Marrow Transplant. 2008, 14,1245–1252).<br />
immuunirekonstituution parantamista on<br />
yritetty manipuloimattomilla luovuttajan<br />
T-soluilla, jolloin riskinä on käänteishyljintä<br />
(GVHD). Hyvä lähestymistapa on ollut tuottaa<br />
antigeenispesi<strong>fi</strong>siä T-soluja. Virusantigeeneina<br />
on käytetty CMV, EBV ja adenovirusta.<br />
Antigeenispesi<strong>fi</strong>sten T-solujen tuottaminen<br />
ei ole uusi asia. se suoritetaan ex<br />
vivo siten, että perifeerisen veren mononukleaarisia<br />
soluja (PBMC) stimuloidaan toistuvasti<br />
antigeenillä, jota esittelee APC (antigen<br />
presenting cell). sen jälkeen soluviljelyssä<br />
monistetaan antigeenispesi<strong>fi</strong>set T-solut<br />
(speci<strong>fi</strong>c CTl). Prosessissa ne T-solut, joilla<br />
on spe<strong>fi</strong>syys muille antigeeneille, eivät säily<br />
(kuva 1).<br />
ebv-CTl<br />
EBV (Epstein-Barr virus) voi aiheuttaa lymfooman<br />
(PTlD, posttransplant lymfoproliferative<br />
disease) kantasolujensiirron jälkeen. Frekvenssi<br />
vaihtelee 1–25 %:n välillä, jos siirre on<br />
rekisteriluovuttajalta (UrD) HlA-epäsopivalta<br />
luovuttajalta (mismatch). siirteen T-soludepleetio<br />
lisää tätä riskiä huomattavasti.<br />
EBV-spesi<strong>fi</strong>siä T-soluja (EBV-CTl) käytettiin<br />
profylaktisesti Helen Heslopin ja työtovereiden<br />
tutkimuksessa (Blood. 2010,<br />
115, 925–935), missä siirteet olivat CD6/<br />
CD8- depletoituja. Potilaista 90 sai CTl-siirron,<br />
eikä kenellekään tullut lymfoomaa, kun<br />
taas kymmenisen prosenttia 42 potilaasta,<br />
jotka eivät saaneet CTl-siirtoa, kehitti lymfoproliferatiivisen<br />
taudin. EBV-CTl oli tehokas<br />
myös PTlD:n hoidossa. Potilaista, joilla<br />
oli aktiivinen lymfoproliferatiivinen tauti,<br />
11/13 saavutti remission, mutta kahdelle ei<br />
tullut vastetta ja he kuolivat. EBV-CTl-siirrot<br />
vähensivät viruskuormaa, paransivat tau-<br />
ASH AsH 2011<br />
43<br />
din 80 %:ssa sekä antoivat virusspesi<strong>fi</strong>sen<br />
immuniteetin ja pitkäaikaisen suojan. Tarvittava<br />
annos oli pieni, 104–105 solua/kg, ja<br />
toksisuus oli alhainen.<br />
TRiviRuS-SPeSiFiSeT T-SoluT<br />
yleisimmät komplikaatioita aiheuttavat<br />
virukset ovat CMV, EBV ja adenovirus, mikä<br />
herätti ajatuksen multispesi<strong>fi</strong>sistä T-soluista.<br />
Useimmat kantasolujen luovuttajat ovat<br />
näille immuuneja, ja heiltä saa hyvin T-soluja.<br />
Näitä kolmea virusta vastaan alettiin tehdä<br />
CTl-solulinjoja käyttäen Ad5f35-vektoria<br />
(Nat Med. 2006, 12, 1160–1166) ja saatiin<br />
aikaan runsaasti solulinjoja, 20 bivirus- ja 36<br />
trivirus-spesi<strong>fi</strong>stä. Paras spesi<strong>fi</strong>syys näytti<br />
olevan CMV:lle.<br />
Trivirus-spesi<strong>fi</strong>sillä CTl-siirroilla oli<br />
profylaktista tehoa. EBV:n suhteen 9/40<br />
potilaasta sai reaktivaation, mutta kaikilla<br />
9:lla viruskuorma laski eikä viruslääkettä<br />
tarvittu. CMV-reaktivaatio kehittyi 10/26:lla<br />
heti CTl-infuusion aikoihin. Näistä 7/10:lla<br />
oli pitkäaikainen teho virukseen, eikä viruslääkettä<br />
tarvittu. yksi potilas kuoli CMV-tautiin.<br />
Adenovirusinfektiota ei tullut kenellekään<br />
29:stä CTl-siirron jälkeen. Kahdeksalla<br />
oli adenovirusta veressä tai ulosteessa, ja<br />
kaikilta virus hävisi 1–2 CTl-annoksen jälkeen.<br />
Kolmella potilaalla oli progressiivinen<br />
adenoviruspneumonia, kaksi heistä hävitti<br />
viruksen CTl:n avulla. latenttia CMV- ja EBVvirusta<br />
vastaan spesi<strong>fi</strong>sten T-solujen todettiin<br />
sekä lisääntyvän että säilyvän potilaassa.<br />
sen sijaan adenospesi<strong>fi</strong>set CTl:t lisääntyivät<br />
vain adenovirusinfektiossa. Trivirusspesi<strong>fi</strong>sillä<br />
T-soluilla saatiin siis hyvä vaste<br />
kaikkiin näihin virusinfektioihin keskimäärin<br />
93 %:ssa.
44<br />
Kuva 1. anTigeenispesiFisTen T-solujen TuoTTaMinen<br />
PBMC<br />
PBMC<br />
IL2<br />
APC<br />
CTL<br />
ANTiGEENi<br />
perifeerisen veren mononukleaariset solut<br />
interleukiini 2<br />
antigeenin esittelevä solu<br />
antigeenispesi<strong>fi</strong>set T-solut<br />
il2<br />
APC<br />
ASH 2011<br />
CTl sPEsiFiNEN<br />
KoHDE-<br />
ANTiGEENillE
Nyt herää tietysti kysymys siitä, miten<br />
näin erinomaiset solut saataisiin kliiniseen<br />
käyttöön. Trivirus-spesi<strong>fi</strong>sten CTl:ien valmistus<br />
on monimutkaista ja kallista. Tarvitaan<br />
GMP-tilat, koulutetut laborantit, CTllinjat<br />
pitää tuottaa ja laatutestaukset suorittaa<br />
ennen kliinistä käyttöä. Ennen kaikkea<br />
prosessi on kovin hidas. Esimerkiksi solulinjan<br />
tuotto vie 4–6 viikkoa, CTl-monistus 3–4<br />
viikkoa, ja laatutestit pari viikkoa. Hintaakin<br />
tulee yhdelle solulinjalle 10 000 yhdysvaltain<br />
dollaria. onkin kehitetty CTl-tuottoon<br />
nopeampi menetelmä, missä virusvektori<br />
jätetään pois ja lähdetään liikkeelle dendriittisolun<br />
nukleofektiosta. Tällä on tuotanto<br />
saatu lyhenemään 13–15 päivään. Erityislaitteistoja<br />
käytetään (Gas permeable rapid<br />
expansion device) nopeampaan soluekspansioon.<br />
rapid CTl:n hinta-arvio oli 3 500<br />
yhdysvaltain dollaria.<br />
CTl-SoluPAnKKi<br />
seuraava askel trivirus-spesi<strong>fi</strong>sten CTl:ien<br />
nopeaan saamiseen on solupankki. solujen<br />
ei tarvitse olla luovuttajaperäisiä, vaan<br />
riittää, että ne ovat riittävässä määrin HlAsopivia<br />
(most closely HlA matched allogeneic<br />
virus-speci<strong>fi</strong>c CTl = CHM-CTl)(Barker,<br />
Blood 2010; Doubrivina, Blood 2011). CTllinjojen<br />
luovuttajina käytettiin henkilöitä,<br />
joilla oli yleisimmät alleelit. Kun kantasolujensiirtopotilaita<br />
seulottiin, sopiva CTl-linja<br />
voitiin identi<strong>fi</strong>oida 68/77:lle. Valtaosalla oli<br />
2-3/6 HlA-sopivuus sekä kantasolujen saajan<br />
että luovuttajan suhteen. yhdysvalloissa<br />
on nyt meneillään monikeskustutkimus,<br />
missä CHM-CTl-infuusioita käytetään kantasolujensiirron<br />
jälkeiseen EBV:n, CMV:n tai<br />
adenoviruksen reaktivaation tai infektion<br />
hoitoon - FDA ei vielä salli profylaktista käyttöä.<br />
Potilaat saavat yhden CHM-CTl-infuusion,<br />
ja jos vaste on osittainen, he voivat<br />
saada kahden viikon välein 4–5 lisäannosta.<br />
jo 48 potilasta on rekrytoitu (23 CMV, 9<br />
EBV, 16 adeno). Hyviä hoitovasteita on ollut,<br />
ASH 2011<br />
45<br />
ja Cr/Pr -frekvenssi on ollut 82 % päivänä<br />
42. Voisi ajatella, että allogeeniset mismatch-T-solut<br />
aiheuttaisivat käänteishyljintää,<br />
mutta näin ei ollut. 33/40 potilaalle ei<br />
tullut mitään haittavaikutuksia, viidelle tuli<br />
gradus i iho-GVHD, ja vain yhdelle gradus iii<br />
maksa-GVHD. Toksisuus on siis katsottava<br />
alhaiseksi. Tulokset ovat kaiken kaikkiaan<br />
varsin lupaavia.<br />
solupankin CTl-linjoilta edellytetään luovuttajien<br />
arviointia ja solulinjojen testausta.<br />
on arvioitu että 22–26 solulinjaa riittäisi kattamaan<br />
HlA-sopivuuden vähintää 3 lokuksessa<br />
noin 75 %:lle kantasolujensiirtopotilaista.<br />
”lasten All:n hoitotulokset<br />
ovat nykyisin erinomaiset,<br />
yli 80 % potilaista<br />
paranee. Tähän on päästy<br />
kehittämällä hoidon<br />
porrastusta riskitekijöihin<br />
perustuen.”<br />
Edinburghissa valmistellaan paraikaa<br />
uutta solupankkia ja optimaalisten luovuttajien<br />
solulinjoja. Voisikohan näitä trivirus-spesi<strong>fi</strong>siä,<br />
HlA-sopivia pankki-CTl-soluja lähivuosina<br />
ostaa kaupasta? se olisi varmaankin<br />
yksinkertaisempaa kuin yrittää alkaa<br />
tuottaa CTl-linjoja suomessa luovuttajan tai<br />
perheenjäsenen (haploidenttisistä) soluista<br />
suomen harvoissa GMP-tiloissa (esim. sPr-<br />
Veripalvelu). Halvasta hoidosta tuskin kuitenkaan<br />
tulee olemaan kyse.
46<br />
lASTen All-RinTAmAn uuSiA TuuliA<br />
lasten All:n hoitotulokset ovat nykyisin<br />
erinomaiset, yli 80 % potilaista paranee.<br />
Tähän on päästy kehittämällä hoidon porrastusta<br />
riskitekijöihin perustuen. Hoitovaste<br />
ja piilevä jäännöstauti ovat näistä määrääviä<br />
nykyprotokollissa perinteisten huonoennusteisten<br />
sytogeneettisten muutosten ohella,<br />
joita ovat erityisesti BCr-ABl1, Mll-geenin<br />
uudelleenjärjestymä sekä hypodiploidia.<br />
Tästäkin huolimatta mitalilla on siis toinen<br />
puoli: 15–20 %:lla tauti uusiutuu, ja sen jälkeen<br />
meneekin paljon huonommin. Hämmentävää<br />
on lisäksi ollut todeta, että noin<br />
puolella relapsipotilaista ovat kaikki tiedetyt<br />
ennustetekijät olleet suotuisia, ja hoitovastekin<br />
on ollut erinomainen. Pohjoismaisissa<br />
NoPHo-protokollissakin tiedämme vanhastaan,<br />
että eniten relapseja on tullut tavallisilla,<br />
”hyvillä” keskiriskipotilailla.<br />
Molekyyligenetiikan runsaudesta nousee<br />
nyt esiin kaksi huomattavaa uutta riskitekijää<br />
B-solulinjan All:ssa, jotka ovat tärkeitä<br />
erityisesti yleisyytensä vuoksi. Nämä ovat<br />
iKAros-geenin muutokset sekä ”BCr-ABl1<br />
like” -ryhmä, joilla myös on päällekkäisyyttä.<br />
iKAros (iKZF1, ikaros zinc <strong>fi</strong>nger 1) on<br />
lymfaattinen transskriptiofaktori, joka edesauttaa<br />
kantasolun kehitystä lymfaattiseksi<br />
prekursoriksi. iKAros-geenin mutaatiot tai<br />
kopiomäärän muutokset johtavat huonoon<br />
ennusteeseen. Ph+ All:ssa iKAros-deleetio<br />
on yli 80 %:ssa, missä sillä on tärkeä osuus<br />
patogeneesissa; ”wild type” iKAros inhiboi<br />
leukemiasolujen proliferaatiota ja aktivoi tuumorisuppressorigeenejä.BCr-ABl1-negatiivisessa<br />
B-prekursori-All:ssa tavataan myös<br />
iKAros-muutoksia. Korkeariski-All:ssa<br />
iKAros-muutoksia on todettu 20–30 %:lla.<br />
Ennusteellinen merkitys korkeariski-All:ssa<br />
on kiistanalainen ja ainakin vähenee, jos<br />
hoito on intensiivinen. Matala- ja keskiriski-<br />
All:ssa yhteensä (vastaa NCi:n standardiriskiä)<br />
iKAros-deleetiota esiintyy vähemmän,<br />
8–14 %. Keskiriski-All:ssa iKAros-muutos-<br />
ASH 2011<br />
ten ennusteellisen merkityksen on jo todettu<br />
olevan suuri yhdysvaltalaisissa ja hollantilaisissa<br />
tutkimuksissa, ja lisää samansuuntaista<br />
tietoa tulee (# 1455 japanista, # 3518<br />
italiasta), iKAros-geenin muutosten ollessa<br />
riippumaton huonon ennusteen merkki.<br />
Tähän iKAros-muutosten ryhmään kuuluvat<br />
myös TEl-AMl1- ja hyperdiploidia-potilaat,<br />
joiden tauteja siis on klassisesti pidetty<br />
hyväennusteisina. iKAros-muutokset näyttävät<br />
johtavan varsinkin myöhäisiin relapseihin.<br />
iKAros-geenin muutoksia kannattaa<br />
siis etsiä B-linjan All:ssa (poissulkien<br />
Ph+ ja Mll-potilaat), erityisesti keskiriskin<br />
All:ssa sisältäen siis myös potilaat, joilla on<br />
TEl-AMl1 tai hyperdiploidia. Tutkimusmenetelmiä<br />
ovat MlPA335 (multiplex ligationdependent<br />
probe ampli<strong>fi</strong>cation) sekä vertaileva<br />
genominen hybridisaatio mikrolevytutkimuksena<br />
(arrayCGH). Tutkimukseen tarvitaan<br />
DNA:ta tai pakastettuja blastisoluja.<br />
”BCr-ABl1-like” (Ph-like) on uusi ryhmä,<br />
joka käyttäytyy geeniekspressioanalyyseissä<br />
kuten Ph+, mutta siinä ei esiinny<br />
Philadelphia-translokaatiota (BCr-ABl1).<br />
Ennuste on yhtä huono kuin Ph+ All:ssa<br />
(EFs 50–60 %). suurimmalla osalla näistä<br />
potilaista on iKZF1-deleetio tai mutaatio.<br />
lähes 50 %:lla potilaista on myös<br />
CrlF2:n (sytokiinireseptorigeeni) uudelleenjärjestymä,<br />
ja n. 30 %:lla jAK-mutaatio.<br />
”BCr-ABl1-like” on varsin yleinen, esiintyen<br />
15–29 %:ssa kaikista B-linjan All:sta<br />
(poissulkien Mll ja Ph+). isolla osalla ei<br />
löydy mitään kromosomaalisia muutoksia.<br />
jos sytogeneettiset tutkimustulokset ovat<br />
ns. normaaleja, noin puolet on osoittautunut<br />
olevan ”BCr-ABl1-like”. CoG:n (# 67)<br />
tutkimuksissa BCr-ABl1-like konstellaatio<br />
löytyi 17 %:lla korkeariskisistä B-linjan<br />
All-potilaista. Genomitutkimuksilla he<br />
osoittivat, että BCr-ABl1-like All:n tyyppilöydöksenä<br />
olivat uudelleenjärjestymät,<br />
mutaatiot ja kopiomäärän muutokset, jotka<br />
dysreguloivat sytokiinireseptorien ja kinaa-
sien signalointia. Toisessa CoG:n työssä<br />
(# 743) tutkittiin selekoimatonta CoG:n<br />
B-prekursori-All -aineistoa käyttäen kahta<br />
eri klusterointialgoritmiä (rosE ja PAM).<br />
BCr-ABl1-like-potilaita löytyi 12–14 %.<br />
Multivarianssianalyyseissä (Cox regressio)<br />
osoittautui, että BCr-ABl1-like oli riippumaton<br />
huonon ennusteen tekijä. Muita huonon<br />
ennusteen tekijöitä olivat tutut MrD ><br />
0.01 % induktion päivänä 29, hypodiploidia<br />
(PAM), sekä näiden lisäksi yli 10 vuoden ikä<br />
ja diagnoosivaiheen perifeerisen veren blastitaso<br />
yli 100000 x 10E9/l (rosE). BCr-<br />
ABl1-like ekspressiota kannattaa hakea<br />
B-linjan All:ssa, varsinkin jos ei löydy kromosomaalisia<br />
muutoksia (”unclassi<strong>fi</strong>ed<br />
ASH 2011<br />
47<br />
B-other”). Tutkimusmenetelmänä on GEP<br />
(geeniekspressiopro<strong>fi</strong>ili), mikä heti asettaa<br />
rajoituksensa. siihen tarvitaan rNA:ta (tai<br />
pakastettuja soluja), eikä GEP ole monessakaan<br />
paikassa vielä kliinisessä rutiinikäytössä,<br />
ei meilläkään.<br />
Ensimmäinen lähtökohta on nyt kehittää<br />
valmiudet tunnistaa nämä potilasryhmät.<br />
sen jälkeen voidaan suunnitella miten<br />
heitä hoidettaisiin. Klassisesti tulee ensiksi<br />
mieleen allogeeninen kantasolujensiirto. on<br />
kuitenkin odotettavissa, että täsmähoitoja<br />
alkaa ilmaantua näköpiiriin, ensimmäisinä<br />
tyrosiinikinaasi-inhibiittorit BCr-ABl1-like<br />
ryhmän potilaille, missä on odotettavissa<br />
tehoa ainakin osalle heistä (# 67).
vesa lindström, HYKS:<br />
KrooNiNEN lyMFAATTiNEN lEUKEMiA (Kll) –<br />
ViHDoiNKiN PiNTAA syVEMMällE?
Kll-solun mikroympäristöstä (microenvironment) on tullut yksi keskeisimmistä<br />
Kll:n tutkimuskohteista viimeisen vuosikymmenen aikana. Tämä<br />
myös näkyi san Diegon AsH 2011 -kokouksen annissa. Kll-solun ja luuytimen<br />
ja/tai imusolmukkeen mikroympäristön välisen signaloinnin on<br />
todettu merkittävästi vaikuttavan mm. Kll:n progressioon, lääkeresistenssin<br />
kehittymiseen ja jäännöstaudin määrään. Keskeisiä tämän interaktion<br />
solutason välittäjiä ovat mesenkymaaliset stroomasolut, monosyyteistä<br />
peräisin olevat ”nurse-likecells” (NlCs) sekä T-solut. lisäksi kemokiinireseptorit,<br />
kuten CXCr4 sekä CXCr5, osallistuvat Kll-solun säätelyyn. Erittäin<br />
tärkeänä signaloinnin välittäjänä pidetään B-solureseptoria (BCr).<br />
BCr-signalointiin vaikuttavien molekyylien esiinmarssi huonoennusteisen<br />
Kll:n hoidossa on alkanut.<br />
Kll-Solun JA miKRoYmPäRiSTön<br />
vuoRovAiKuTuKSeSTA<br />
Tämänhetkisen käsityksen Kll-solun ja<br />
mikroympäristön välisestä vuorovaikutuksesta<br />
esitti jan Burger MD Anderson Cancer<br />
Centeristä. Kll-solun kontakti NlC-solujen<br />
tai mesenkymaalisten stroomasolujen<br />
välillä muodostuu Kll-solun pinnalla olevien<br />
kemokiinireseptoreiden ja adheesiomolekyylien<br />
välityksellä. NlC-solut suojaavat<br />
Kll-soluja apoptoosilta ja myös lääkehoidon<br />
aiheuttamalta solukuolemalta. Kll-potilaalta<br />
NlC-soluja löytyy pernasta sekä imukudoksesta.<br />
Kll- ja NlC -solujen välisiä reaktioita<br />
koskevat tutkimukset ovat tuottaneet<br />
tärkeää tietoa Kll-solujen mikroympäristöstä<br />
ja avanneet mahdollisuuksia uusien hoitomuotojen<br />
kehittämiseen.<br />
Kll-solujen pinnalla on erityisen runsaasti<br />
CXCr4 ja CXCr5 -kemokiinireseptoreja.<br />
NlC-solut sekä mesenkymaaliset<br />
stroomasolut houkuttelevat Kll-soluja näiden<br />
G-proteiiniin liittyneiden reseptoreiden<br />
kautta. Kll-solujen CXCr4-reseptori voidaan<br />
blokata CXCr4-antagonistin, pleriksaforin,<br />
avulla. rituksi-mabi-pleriksafori -kombinaatiohoidolla<br />
tehtävä kliininen tutkimus<br />
relapsoituneilla Kll-potilailla on meneillään.<br />
CXCr4-antagonistilla Kll-solut mobilisoidaan<br />
kudoksista rituksimabin ulottuville.<br />
ASH AsH 2011<br />
49<br />
Alustavat tulokset tutkimuksesta ovat osoittaneet<br />
pleriksaforin mobilisoivan Kll-soluja<br />
annosriippuvaisesti kudoksista perifeeriseen<br />
vereen.<br />
T-lymfosyyttien merkitys Kll:n progressioon<br />
ja taudinkuvaan on monitahoinen.<br />
Hoitamattomilla Kll-potilailla T-lymfosyyttien<br />
määrä on lisääntynyt, minkä arvellaan<br />
johtuvan vuorovaikutuksesta joko Kll-solujen<br />
tai Kll-potilailla yleisesti esiintyvien<br />
mikrobiantigeenien kanssa. T-lymfosyyttejä<br />
on etenkin luuytimen ja imusolmukkeiden<br />
proliferaatiokeskuksissa, joissa tapahtuu<br />
Kll-solujen nopea jakaantuminen. T-lymfosyyttien<br />
toiminta on myös häiriintynyt johtaen<br />
Kll ja T -solujen välisten immunologisten<br />
reaktioiden häiriöihin. T-lymfosyytit<br />
voivat joko heikentää tai lisätä Kll-kloonin<br />
kasvua. Aktivoitunut Kll-solu tuottaa<br />
T-soluja houkuttelevia CCl3 ja CCl4 -kemokiineja<br />
BCr-stimulaation välityksellä. Korkeiden<br />
CCl3 ja CCl4 -tasojen on todettu liittyvän<br />
nopeampaan Kll:n etenemiseen hoitoa<br />
vaativaksi.<br />
KinAASi-inHibiiTToRiT TuloSSA Kll:n<br />
HoiToon<br />
sopivassa mikroympäristössä Kll-solujen<br />
B-solureseptorit aktivoituvat autoantigeenien<br />
vaikutuksesta johtaen Kll-solun pro-
50<br />
liferaatioon. Tämän antigeenistimulaation<br />
ja BCr-aktivaation on todettu olevan merkittävässä<br />
roolissa Kll:n patogeneesissä ja<br />
ennusteessa (kuva 1). Kll:n ennuste näyttäisi<br />
korreloivan eri BCr-alueilla esiintyvien<br />
somaattisten mutaatioiden määrään. BCrstimulaatio<br />
saa aikaan syk-, Btk- ja Pi3Kδ<br />
-tyrosiinikinaasien aktivaation ja erityisesti<br />
ei-mutatoituneet (unmutated) ja/tai ZAP-<br />
70+Kll-potilaat reagoivat tähän BCr-stimulaatioon.<br />
Näin ollen juuri huonon ennusteen<br />
Kll-potilaiden hoitoon odotetaan tehoa<br />
uusista B-solureseptoriin liittyvien kinaasien<br />
inhibiittoreista. Näistä uusista inhibiittoreista<br />
pisimmällä tutkimuksissa ovat syk<br />
(spleentyrosinekinase)-inhibiittori fostamatinibi,<br />
Btk (Bruton tyrosinekinase)-inhibiittori<br />
PCi-32765 sekä Pi3Kδ (fosfatidylinositoli-3-kinase)-inhibiittori<br />
CAl-101.Näillä<br />
inhibiittoreilla hoidetuilla relapsoituneilla,<br />
Kuva 1. Kll-solun MiKroyMpärisTö<br />
CCl3<br />
bCR<br />
ANTiGEENi<br />
T-solUT<br />
CD40<br />
sElViyTyMiNEN &<br />
ProliFErAATio<br />
Btk<br />
Pl3Ks<br />
CD79<br />
a,b<br />
syk<br />
Kll<br />
nCl<br />
CD40l<br />
liiKKUVUUs &<br />
PysyVyys<br />
CXCl12,<br />
CXCl13<br />
ASH 2011<br />
refraktaareilla Kll-potilailla on tyypillisesti<br />
nähty ensimmäisten hoitoviikkojen/-kuukausien<br />
aikana väliaikaisesti lisääntyvää lymfosytoosia<br />
johtuen Kll-solujen mobilisoinnista<br />
verenkiertoon sekä nopeaa imusolmukkeiden<br />
pienenemistä tai jopa lymfadenopatian<br />
häviämistä. lymfosytoosin on todettu korjaantuvan<br />
puolen vuoden jatkuvan hoidon<br />
jälkeen. Näiltä uusilta hyvin lupaavilta aineilta<br />
puuttuu myös luuydintä lamaava vaikutus.<br />
Kll:n hoidon odotetaan jopa mullistuvan<br />
uusien B-solureseptoriin liittyvien kinaasi-inhibiittorien<br />
ansiosta.<br />
PCi-32765 on suun kautta otettava<br />
Btk-inhibiittori, jonka aikaisemmassa faasi<br />
ib/ii -tutkimuksessa (Byrd, AsCo 2011)<br />
on osoitettu olevan tehokas ja hyvin siedetty<br />
Kll:n hoidossa. Nyt julkistettiin tämän<br />
monikeskustutkimuksen pitkäaikaisseurannan<br />
tulokset (# 983). Päivittäin annostel-<br />
syk<br />
Btk<br />
g<br />
Pl3Ks<br />
CXCr4,<br />
CXCr5<br />
VCAM-1, FN<br />
CD49d<br />
(VlA-4)<br />
CXCl12<br />
mSC
tavalla PCi-32765 -inhibiittorilla hoidettiin<br />
kaksi Kll-kohorttia, aikaisemmin hoitamattomat<br />
≥ 65-vuotiaat sekä vähintään kahden<br />
hoidon jälkeen relapsoituneet tai refraktaarit<br />
tapaukset. inhibiittoria annosteltiin 28<br />
päivän sykleissä, kunnes Kll progredioi.<br />
Annoksia oli kaksi, 420 mg päivässä molemmissa<br />
kohorteissa sekä 840 mg päivittäin<br />
relapsoituneilla/refraktaareilla potilailla.<br />
Tuloksia kerrottiin nyt relapsoitu-neen/refraktaarin<br />
-kohortin osalta. Tämä kohortti<br />
käsitti 61 potilasta, 27 potilasta sai 420 mg<br />
päivässä ja 34 potilasta sai 840 mg. Keskimääräinen<br />
seuranta-aika 420 mg saaneista<br />
oli 10,2 kuukautta ja 840 mg saaneista<br />
6,5 kuukautta. 72 %:lla oli vähintään yhden<br />
huonon ennusteen biologinen markkeri<br />
(del17p 31 %, del11q 33 %, igVHunmutated<br />
57 %). Tunnusomainen vasteen merkki,<br />
väliaikainen lymfosytoosi seurannassa<br />
väistyen, todettiin suurimmalla osalla potilaista.<br />
iWCll-kriteereiden mukainen vaste<br />
(orr; Pr + Cr) 420 mg saaneista todettiin<br />
jopa 70 %:lla potilaista (aikaisemmassa faasi<br />
ib/ii -tutkimuksessa 48 %:lla). orr 840<br />
mg päivittäin saaneista oli seuranta-ajan jälkeen<br />
44 %. Vasteen saaminen näytti olevan<br />
riippumatonta ennusteellisista markkereista.<br />
Tavallisimmat gradus 1–2 haittavaikutukset<br />
olivat ripuli, väsymys, pahoinvointi<br />
sekä mustelmat. Vakavia inhibiittoriin liittyviä<br />
haittavaikutuksia esiintyi 10 %:lla.<br />
82 %:a tutkimukseen osallistuneista jatkaa<br />
edelleen inhibiittorin käyttöä ja vain 8 %:lla<br />
tauti on progredioinut (5/61 potilasta). Kuuden<br />
kuukauden PFs 420 mg saaneista oli 92<br />
% ja 840 mg saaneista 90 %. Faasi iii -tutkimus<br />
on suunnitteilla.<br />
CAl-101 on suun kautta otettava spesi<strong>fi</strong>nen<br />
fosfatidylinositoli-3-kinaasin (Pi3Kδ)<br />
estäjä, joka on edennyt jo kombinaatiohoitotutkimukseen.<br />
Aikaisemmassa faasi i -tutkimuksessa<br />
CAl-101-monoterapia osoittautui<br />
tehokkaaksi Kll-potilailla (Blood<br />
2010;116:55). Tässä kokouksessa julkaistiin<br />
faasi i -tutkimus (# 1787), jossa CAl-<br />
101 (Gs-1101) kombinoitiin rituksimabiin<br />
ja/tai bendamustiiniinrelapoi-tuneilla/refraktaareilla<br />
Kll-potilailla. 27 aikaisemmin<br />
hoidettua Kll-potilasta jakautui kolmeen eri<br />
tutkimushaaraan. 14 potilasta sai Gs-1101<br />
yhdistettynä rituksimabiin (Gr), kymmenen<br />
Gs-1101 yhdistettynä bendamustiiniin (GB)<br />
ASH 2011<br />
51<br />
ja kolme Gs-1101 yhdistettynä molempiin<br />
(GrB). Gs-1101-annos vaihteli 100–150mg<br />
kahdesti päivässä Gr- ja GB -ryhmissä ja<br />
GrB-ryhmän kolme potilasta saivat 150 mg<br />
kahdesti päivässä. Gr-ryhmän rituksimabiannos<br />
oli 375 mg/m 2 viikoittain annettuna<br />
yhteensä kahdeksan annosta. GB-ryhmässäbendamustiinia<br />
annettiin 90 mg/m 2<br />
päivinä 1 ja 2 yhteensä kuusi sykliä, syklin<br />
pituus oli 28 vrk. GrB-ryhmässärituksimabia<br />
potilaat saivat 375 mg/m 2 syklin päivä-<br />
”juuri huonon ennusteen<br />
Kll-potilaiden hoitoon<br />
odotetaan tehoa uusista<br />
B-solureseptoriin liittyvien<br />
kinaasien inhibiittoreista.”<br />
nä 1 yhteensä kuusi sykliä, bendamustiinia<br />
annettiin kuten GB-ryhmässä. suurin osa<br />
potilaista oli yli 60-vuotiaita, lymfadenopatia<br />
oli laajaalaista ja lähes kaikki olivat aikaisempina<br />
hoitoina jo saaneet rituksimabia tai<br />
bendamustiinia tai molempia. Vasteet olivat<br />
huomattavia, lymfadenopatia väheni yli puoleen<br />
jokaisessa ryhmässä yli 75 %:lla potilaista.<br />
imusolmukkeiden pieneneminen oli<br />
nopeaa ja havaittavissa enintään 2 syk-lin<br />
jälkeen. Hoidon siedettävyydessä ei tullut<br />
yllätyksiä. Gs-1101-pohjaisen kombinaatiohoidon<br />
todettiin olevan hyvin tehokas lymfadenopatiaan<br />
ja tämän tutkimuksen pohjalta<br />
on suunnitteilla jo faasi iii -tutkimus. rekrytointitutkimus,<br />
jossa Gs-1101 on kombinoitu<br />
fludarabiiniin tai ofatumumabiin on myös<br />
jo meneillään.<br />
AlemTuTSumAbi JA lenAlidomidi<br />
– nYKYnäKemYKSeT Kll:n<br />
HoiTovAiHToeHToinA<br />
Noin 7 %:lla aikaisemmin hoitamattomista<br />
Kll-potilaista löydetään 17p-deleetio tai<br />
p53-mutaatio. Kuten hyvin tiedämme, nämä<br />
hyvin huonon ennusteen Kll-potilaat ovat
52<br />
resistenttejä sytostaattihoidoille. Cll8-tutkimuksessa<br />
Cr-tason vaste näillä potilailla<br />
saavutettiin FCr-hoidolla vain noin 5 %:lla.<br />
lisäksi keskimääräinen aika ennen taudin<br />
etenemistä (PFs) 17p-deleetio potilailla oli<br />
vain 11,2 kuukautta. Kombinaatiohoidosta,<br />
jossa alemtuzumabi on yhdistettynä korkeaannossteroidiin,<br />
on viimeaikaisissa tutkimuksissa<br />
osoitettu olevan tehoa. Kaikki 17<br />
aikaisemmin hoitamatonta 17p-deleetion<br />
omaavaa Kll-potilasta saivat vasteen Cam-<br />
Pred-hoidolle, jossa alemtuzumabi yhdistettiin<br />
korkea-annosmetyyliprednisoloniin.<br />
65 % sai täyden vasteen. Vain 14 % vastasi<br />
hoidolle, jos CamPred-hoito ei ollut ensi<br />
linjan hoitona. (leukemia 2006;20:1441–<br />
1445). Toisessa tutkimuksessa alemtuzumabi<br />
yh-distettiin deksametasoniin (Cam-<br />
Dex), mutta vasteet eivät olleet aivan yhtä<br />
hyviä kuin CamPred-hoidolla on saatu<br />
aikaan (Blood 2010;116:920). Peter Hillmen<br />
omassa puheenvuorossaan ottikin vahvan<br />
kannan CamPred-hoidon puolesta tälle huonon<br />
ennusteen Kll-potilasryhmälle. soveltuvat<br />
potilaat tulisi ohjata myös allogeeniseen<br />
kantasolujensiirtoon.<br />
lenalidomidin osalta tämän kokouksen<br />
antia olivat yhdistelmähoitotutkimukset.<br />
Badoux ym. MD Anderson -keskuksesta<br />
(# 642) julkaisivat lopulliset tulokset faasi ii<br />
-tutkimuksesta, jossa lenalidomidi yhdis-tettiin<br />
rituksimabiin relapsoituneilla/refraktaareilla<br />
Kll-potilailla. Kaikki 59 potilasta olivat<br />
saaneet aikaisemmin puriinianalogi-pohjaista<br />
hoitoa. lenalidomidia annettiin 10 mg päivässä<br />
28 päivän sykleissä alkaen syklin 1 päivästä<br />
9. rituksimabia annettiin 375 mg/m2<br />
viikoittain neljän viikon ajan 1.syklin päivästä<br />
1 alkaen ja tämän jälkeen syklin päivänä<br />
1 kaikkiaan 12 sykliin saakka. lenalidomidia<br />
annettiin 12 kuukauden ajan, mutta mikäli<br />
vastetta saatiin, sen käyttöä voitiin jatkaa.<br />
Potilaiden keski-ikä oli 64 vuotta ja 98 % oli<br />
saanut aikaisemmin rituksimabihoitoa. Vaste<br />
arvioitiin 3 ja 6 syklin jälkeen ja sittemmin<br />
jokaisen 6 syklin jälkeen. Vastetta (orr) saatiin<br />
66 %:lle potilaista, täyden vasteen (Cr)<br />
sai 6 potilasta (10 %), osittaisen vasteen<br />
ASH 2011<br />
imusolmukkeisiin 10 potilasta (nPr, 17 %)<br />
ja Pr-tason vasteen sai 23 potilasta (39 %).<br />
15 potilaalla oli 17p-deleetio. Tässä ryhmässä<br />
kaikkiaan vastetta tuli 53 %:lle, Cr-vasteen<br />
sai kaksi potilasta (13 %), nPr-vasteen<br />
samoin kaksi potilasta ja Pr-vasteen saavutti<br />
neljä potilasta (27 %). 25 kuukauden keskimääräisen<br />
seuranta-ajan jälkeen 15 potilasta<br />
(25 %) on edelleen jatkanut hoitoa. Elossa<br />
on 49 potilasta ja kahden vuoden os (overall<br />
survival) on peräti 83 %. Gradus 3–4 neutropenia<br />
ilmaantui 40 potilaalle (47 %) ja trombopenia<br />
13 potilaalle (22 %). Vakavan infektion<br />
sai kahdeksan potilasta (31 %). lenalidomidi-rituksimabi<br />
-kombinaatiohoidolla saatiin<br />
siis kestävää vastetta tässä hoidollisesti<br />
haastavassa ryhmässä, vastetta tuli myös<br />
17p-deleetiopotilaille.<br />
JäännöSTAudin meRKiTYS<br />
Boettcher ja kumppanit seurasivat saksan<br />
Kll-ryhmän Cll8-tutkimukseen osallistuneiden<br />
jäännöstaudin (MrD) kehittymistä ja<br />
ennusteellista merkitystä (# 1777). jäännöstautia<br />
seurattiin hoitotauon aikana 256 potilaalla,<br />
joiden Kll ei edennyt vuoteen hoitojen<br />
päättymisestä. Keskimääräinen jäännöstaudin<br />
kasvu oli 6,3-kertainen seuranta-aikana<br />
koko ryhmässä. 25 % potilaista pysyi MrDnegatiivisina<br />
koko seuranta-ajan. 37 %:lla<br />
potilaista MrD jäi mediaania pienemmäksi<br />
(ryhmä 2) ja 39 %:lla jäännöstaudin määrä<br />
lisääntyi yli mediaanin (ryhmä 3). Tässä ryhmässä,<br />
jossa MrD kasvoi nopeammin, aika<br />
Kll:n etenemiseen (PFs) jäi lyhyemmäksi<br />
(40 kuukautta) verrattuna ryhmään 2 (PFs<br />
66 kuukautta). ryhmässä 2 oli 2,5 kertaa<br />
pienempi riski Kll:n kliiniseen etenemiseen<br />
kuin ryhmässä 3. MrD:n itsenäinen merkitys<br />
Kll:n etenemiseen säilyi myös monimuuttuja-analyysissä.<br />
Kirjoittajat ennustavatkin<br />
ylläpitohoidon olevan tehokkain potilaille,<br />
jotka vastaavat hoitoon, mutta relapsoituvat<br />
aikaisemmin jäännöstaudin nopeamman<br />
lisääntymisen takia. ylläpitohoitostrategiat<br />
voivat olla arkipäivää myös Kll:n hoidossa<br />
tulevaisuudessa follikulaarisen lymfooman<br />
hoidon kaltaisesti.
<strong>fi</strong>1202024479