

A Toy Model of Chemical Reaction Networks - TBI - Universität Wien
A Toy Model of Chemical Reaction Networks - TBI - Universität Wien
A Toy Model of Chemical Reaction Networks - TBI - Universität Wien

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
3.5. PERFORMANCE 31<br />
3.5 Performance<br />
In order to validate the energy calculation, predicted total atomization energies<br />
(TAE) have been compared to experimental ones, see figs. 3.9 and 3.10.<br />
Experimental TAE values are taken from [18].<br />
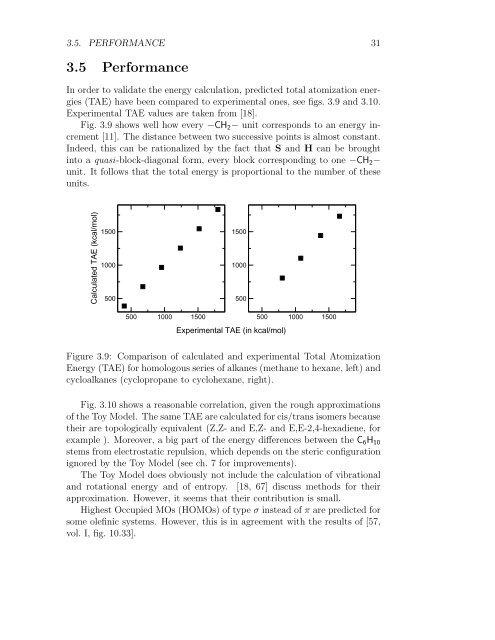
Fig. 3.9 shows well how every −CH 2 − unit corresponds to an energy increment<br />
[11]. The distance between two successive points is almost constant.<br />
Indeed, this can be rationalized by the fact that S and H can be brought<br />
into a quasi-block-diagonal form, every block corresponding to one −CH 2 −<br />
unit. It follows that the total energy is proportional to the number <strong>of</strong> these<br />
units.<br />
Calculated TAE (kcal/mol)<br />
1500<br />
1000<br />
500<br />
1500<br />
1000<br />
500<br />
500 1000 1500<br />
500 1000 1500<br />
Experimental TAE (in kcal/mol)<br />
Figure 3.9: Comparison <strong>of</strong> calculated and experimental Total Atomization<br />
Energy (TAE) for homologous series <strong>of</strong> alkanes (methane to hexane, left) and<br />
cycloalkanes (cyclopropane to cyclohexane, right).<br />
Fig. 3.10 shows a reasonable correlation, given the rough approximations<br />
<strong>of</strong> the <strong>Toy</strong> <strong>Model</strong>. The same TAE are calculated for cis/trans isomers because<br />
their are topologically equivalent (Z,Z- and E,Z- and E,E-2,4-hexadiene, for<br />
example ). Moreover, a big part <strong>of</strong> the energy differences between the C 6 H 10<br />
stems from electrostatic repulsion, which depends on the steric configuration<br />
ignored by the <strong>Toy</strong> <strong>Model</strong> (see ch. 7 for improvements).<br />
The <strong>Toy</strong> <strong>Model</strong> does obviously not include the calculation <strong>of</strong> vibrational<br />
and rotational energy and <strong>of</strong> entropy. [18, 67] discuss methods for their<br />
approximation. However, it seems that their contribution is small.<br />
Highest Occupied MOs (HOMOs) <strong>of</strong> type σ instead <strong>of</strong> π are predicted for<br />
some olefinic systems. However, this is in agreement with the results <strong>of</strong> [57,<br />
vol. I, fig. 10.33].













