


You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Furthermore, the <strong>in</strong>itial models for the aspartic prote<strong>in</strong>ases ren<strong>in</strong> and ren<strong>in</strong>-<strong>in</strong>hibitor complexes were built by us<strong>in</strong>g<br />
the 3D structure of the remotely related fungal prote<strong>in</strong>ases [60-62]. Later on, the models for ren<strong>in</strong> were constructed<br />
by employ<strong>in</strong>g the structures of mammalian aspartic proteases, peps<strong>in</strong> and chymos<strong>in</strong> [63,64]. Comparative analysis of<br />
fungal and mammalian ren<strong>in</strong> models revealed that the <strong>in</strong>accuracies <strong>in</strong> the models occurred due to the dissimilarity <strong>in</strong> the<br />
arrangement of helices and strands between the fungal and mammalian prote<strong>in</strong>ases, as well as the slightly different variable<br />
regions. On the other hand, the model<strong>in</strong>g of ren<strong>in</strong> active site was reasonably accurate [65].<br />
Early <strong>in</strong> the eighties, manual homology model<strong>in</strong>g was assisted by manoeuvr<strong>in</strong>g of prote<strong>in</strong> molecules on the graphics<br />
term<strong>in</strong>al that was made achievable by computer programs like FRODO [66]. S<strong>in</strong>ce then, many different homology<br />
model<strong>in</strong>g packages have been developed [42], which can be grouped <strong>in</strong>to three different groups: rigid-body assembly,<br />
segment match<strong>in</strong>g, or model<strong>in</strong>g by satisfaction of spatial restra<strong>in</strong>ts [67].<br />
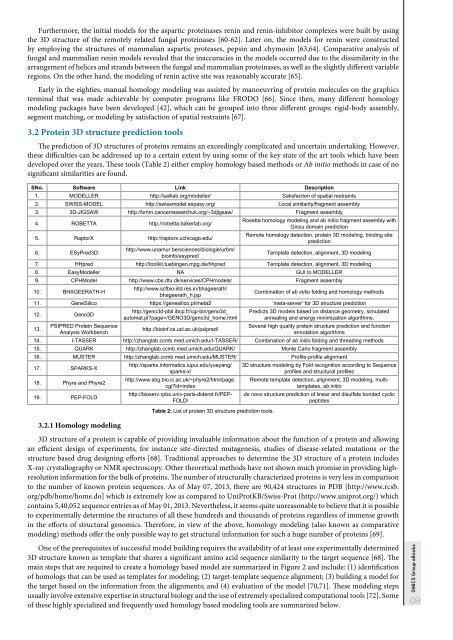
3.2 Prote<strong>in</strong> 3D structure prediction tools<br />
The prediction of 3D structures of prote<strong>in</strong>s rema<strong>in</strong>s an exceed<strong>in</strong>gly complicated and uncerta<strong>in</strong> undertak<strong>in</strong>g. However,<br />
these difficulties can be addressed up to a certa<strong>in</strong> extent by us<strong>in</strong>g some of the key state of the art tools which have been<br />
developed over the years. These tools (Table 2) either employ homology based methods or Ab <strong>in</strong>itio methods <strong>in</strong> case of no<br />
significant similarities are found.<br />
SNo. Software L<strong>in</strong>k Description<br />
1. MODELLER http://salilab.org/modeller/ Satisfaction of spatial restra<strong>in</strong>ts<br />
2. SWISS-MODEL http://swissmodel.expasy.org/ Local similarity/fragment assembly<br />
3. 3D-JIGSAW http://bmm.cancerresearchuk.org/~3djigsaw/ Fragment assembly<br />
4. ROBETTA http://robetta.bakerlab.org/<br />
5. RaptorX http://raptorx.uchicago.edu/<br />
6. ESyPred3D<br />
http://www.unamur.be/sciences/biologie/urbm/<br />
bio<strong>in</strong>fo/esypred/<br />
Rosetta homology model<strong>in</strong>g and ab <strong>in</strong>itio fragment assembly with<br />
G<strong>in</strong>zu doma<strong>in</strong> prediction<br />
Remote homology detection, prote<strong>in</strong> 3D model<strong>in</strong>g, b<strong>in</strong>d<strong>in</strong>g site<br />
prediction<br />
Template detection, alignment, 3D model<strong>in</strong>g<br />
7. HHpred http://toolkit.tueb<strong>in</strong>gen.mpg.de/hhpred Template detection, alignment, 3D model<strong>in</strong>g<br />
8. EasyModeller NA GUI to MODELLER<br />
9. CPHModel http://www.cbs.dtu.dk/services/CPHmodels/ Fragment assembly<br />
10. BHAGEERATH-H<br />
http://www.scfbio-iitd.res.<strong>in</strong>/bhageerath/<br />
bhageerath_h.jsp<br />
Comb<strong>in</strong>ation of ab <strong>in</strong>itio fold<strong>in</strong>g and homology methods<br />
11. GeneSilico https://genesilico.pl/meta2 ‘meta-server’ for 3D structure prediction<br />
12. Geno3D<br />
13.<br />
PSIPRED Prote<strong>in</strong> Sequence<br />
Analysis Workbench<br />
http://geno3d-pbil.ibcp.fr/cgi-b<strong>in</strong>/geno3d_<br />
automat.pl?page=/GENO3D/geno3d_home.html<br />
http://bio<strong>in</strong>f.cs.ucl.ac.uk/psipred/<br />
Predicts 3D models based on distance geometry, simulated<br />
anneal<strong>in</strong>g and energy m<strong>in</strong>imization algorithms.<br />
Several high quality prote<strong>in</strong> structure prediction and function<br />
annotation algorithms<br />
14. I-TASSER http://zhanglab.ccmb.med.umich.edu/I-TASSER/ Comb<strong>in</strong>ation of ab <strong>in</strong>itio fold<strong>in</strong>g and thread<strong>in</strong>g methods<br />
15. QUARK http://zhanglab.ccmb.med.umich.edu/QUARK/ Monte Carlo fragment assembly<br />
16. MUSTER http://zhanglab.ccmb.med.umich.edu/MUSTER/ Profile-profile alignment<br />
17. SPARKS-X<br />
18. Phyre and Phyre2<br />
19. PEP-FOLD<br />
3.2.1 Homology model<strong>in</strong>g<br />
http://sparks.<strong>in</strong>formatics.iupui.edu/yueyang/<br />
sparks-x/<br />
http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.<br />
cgi?id=<strong>in</strong>dex<br />
http://bioserv.rpbs.univ-paris-diderot.fr/PEP-<br />
FOLD/<br />
Table 2: List of prote<strong>in</strong> 3D structure prediction tools.<br />
3D structure model<strong>in</strong>g by Fold recognition accord<strong>in</strong>g to Sequence<br />
profiles and structural profiles<br />
Remote template detection, alignment, 3D model<strong>in</strong>g, multitemplates,<br />
ab <strong>in</strong>itio<br />
de novo structure prediction of l<strong>in</strong>ear and disulfide bonded cyclic<br />
peptides<br />
3D structure of a prote<strong>in</strong> is capable of provid<strong>in</strong>g <strong>in</strong>valuable <strong>in</strong>formation about the function of a prote<strong>in</strong> and allow<strong>in</strong>g<br />
an efficient design of experiments, for <strong>in</strong>stance site-directed mutagenesis, studies of disease-related mutations or the<br />
structure based drug design<strong>in</strong>g efforts [68]. Traditional approaches to determ<strong>in</strong>e the 3D structure of a prote<strong>in</strong> <strong>in</strong>cludes<br />
X-ray crystallography or NMR spectroscopy. Other theoretical methods have not shown much promise <strong>in</strong> provid<strong>in</strong>g highresolution<br />
<strong>in</strong>formation for the bulk of prote<strong>in</strong>s. The number of structurally characterized prote<strong>in</strong>s is very less <strong>in</strong> comparison<br />
to the number of known prote<strong>in</strong> sequences. As of May 07, 2013, there are 90,424 structures <strong>in</strong> PDB [http://www.rcsb.<br />
org/pdb/home/home.do] which is extremely low as compared to UniProtKB/Swiss-Prot (http://www.uniprot.org/) which<br />
conta<strong>in</strong>s 5,40,052 sequence entries as of May 01, 2013. Nevertheless, it seems quite unreasonable to believe that it is possible<br />
to experimentally determ<strong>in</strong>e the structures of all these hundreds and thousands of prote<strong>in</strong>s regardless of immense growth<br />
<strong>in</strong> the efforts of structural genomics. Therefore, <strong>in</strong> view of the above, homology model<strong>in</strong>g (also known as comparative<br />
model<strong>in</strong>g) methods offer the only possible way to get structural <strong>in</strong>formation for such a huge number of prote<strong>in</strong>s [69].<br />
One of the prerequisites of successful model build<strong>in</strong>g requires the availability of at least one experimentally determ<strong>in</strong>ed<br />
3D structure known as template that shares a significant am<strong>in</strong>o acid sequence similarity to the target sequence [68]. The<br />
ma<strong>in</strong> steps that are required to create a homology based model are summarized <strong>in</strong> Figure 2 and <strong>in</strong>clude: (1) identification<br />
of homologs that can be used as templates for model<strong>in</strong>g; (2) target-template sequence alignment; (3) build<strong>in</strong>g a model for<br />
the target based on the <strong>in</strong>formation from the alignments; and (4) evaluation of the model [70,71]. These model<strong>in</strong>g steps<br />
usually <strong>in</strong>volve extensive expertise <strong>in</strong> structural biology and the use of extremely specialized computational tools [72]. Some<br />
of these highly specialized and frequently used homology based model<strong>in</strong>g tools are summarized below.<br />
OMICS Group eBooks<br />
09
