

Initial sequencing and analysis of the human genome - Vitagenes
Initial sequencing and analysis of the human genome - Vitagenes
Initial sequencing and analysis of the human genome - Vitagenes

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
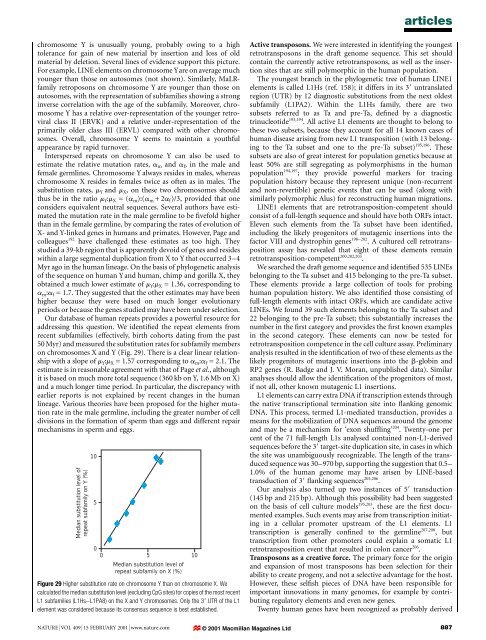
articleschromosome Y is unusually young, probably owing to a hightolerance for gain <strong>of</strong> new material by insertion <strong>and</strong> loss <strong>of</strong> oldmaterial by deletion. Several lines <strong>of</strong> evidence support this picture.For example, LINE elements on chromosome Yare on average muchyounger than those on autosomes (not shown). Similarly, MaLRfamilyretroposons on chromosome Y are younger than those onautosomes, with <strong>the</strong> representation <strong>of</strong> subfamilies showing a stronginverse correlation with <strong>the</strong> age <strong>of</strong> <strong>the</strong> subfamily. Moreover, chromosomeY has a relative over-representation <strong>of</strong> <strong>the</strong> younger retroviralclass II (ERVK) <strong>and</strong> a relative under-representation <strong>of</strong> <strong>the</strong>primarily older class III (ERVL) compared with o<strong>the</strong>r chromosomes.Overall, chromosome Y seems to maintain a youthfulappearance by rapid turnover.Interspersed repeats on chromosome Y can also be used toestimate <strong>the</strong> relative mutation rates, a m <strong>and</strong> a f , in <strong>the</strong> male <strong>and</strong>female germlines. Chromosome Y always resides in males, whereaschromosome X resides in females twice as <strong>of</strong>ten as in males. Thesubstitution rates, m Y <strong>and</strong> m X , on <strong>the</strong>se two chromosomes shouldthus be in <strong>the</strong> ratio m Y :m X =(a m ):(a m +2a f )/3, provided that oneconsiders equivalent neutral sequences. Several authors have estimated<strong>the</strong> mutation rate in <strong>the</strong> male germline to be ®vefold higherthan in <strong>the</strong> female germline, by comparing <strong>the</strong> rates <strong>of</strong> evolution <strong>of</strong>X- <strong>and</strong> Y-linked genes in <strong>human</strong>s <strong>and</strong> primates. However, Page <strong>and</strong>colleagues 192 have challenged <strong>the</strong>se estimates as too high. Theystudied a 39-kb region that is apparently devoid <strong>of</strong> genes <strong>and</strong> resideswithin a large segmental duplication from X to Y that occurred 3±4Myr ago in <strong>the</strong> <strong>human</strong> lineage. On <strong>the</strong> basis <strong>of</strong> phylogenetic <strong>analysis</strong><strong>of</strong> <strong>the</strong> sequence on <strong>human</strong> Y<strong>and</strong> <strong>human</strong>, chimp <strong>and</strong> gorilla X, <strong>the</strong>yobtained a much lower estimate <strong>of</strong> m Y :m X = 1.36, corresponding toa m :a f = 1.7. They suggested that <strong>the</strong> o<strong>the</strong>r estimates may have beenhigher because <strong>the</strong>y were based on much longer evolutionaryperiods or because <strong>the</strong> genes studied may have been under selection.Our database <strong>of</strong> <strong>human</strong> repeats provides a powerful resource foraddressing this question. We identi®ed <strong>the</strong> repeat elements fromrecent subfamilies (effectively, birth cohorts dating from <strong>the</strong> past50 Myr) <strong>and</strong> measured <strong>the</strong> substitution rates for subfamily memberson chromosomes X <strong>and</strong> Y (Fig. 29). There is a clear linear relationshipwith a slope <strong>of</strong> m Y :m X = 1.57 corresponding to a m :a f = 2.1. Theestimate is in reasonable agreement with that <strong>of</strong> Page et al., althoughit is based on much more total sequence (360 kb on Y, 1.6 Mb on X)<strong>and</strong> a much longer time period. In particular, <strong>the</strong> discrepancy wi<strong>the</strong>arlier reports is not explained by recent changes in <strong>the</strong> <strong>human</strong>lineage. Various <strong>the</strong>ories have been proposed for <strong>the</strong> higher mutationrate in <strong>the</strong> male germline, including <strong>the</strong> greater number <strong>of</strong> celldivisions in <strong>the</strong> formation <strong>of</strong> sperm than eggs <strong>and</strong> different repairmechanisms in sperm <strong>and</strong> eggs.Median substitution level <strong>of</strong>repeat subfamily on Y (%)10500 5 10Median substitution level <strong>of</strong>repeat subfamily on X (%)Figure 29 Higher substitution rate on chromosome Y than on chromosome X. Wecalculated <strong>the</strong> median substitution level (excluding CpG sites) for copies <strong>of</strong> <strong>the</strong> most recentL1 subfamilies (L1Hs±L1PA8) on <strong>the</strong> X <strong>and</strong> Y chromosomes. Only <strong>the</strong> 39 UTR <strong>of</strong> <strong>the</strong> L1element was considered because its consensus sequence is best established.Active transposons. We were interested in identifying <strong>the</strong> youngestretrotransposons in <strong>the</strong> draft <strong>genome</strong> sequence. This set shouldcontain <strong>the</strong> currently active retrotransposons, as well as <strong>the</strong> insertionsites that are still polymorphic in <strong>the</strong> <strong>human</strong> population.The youngest branch in <strong>the</strong> phylogenetic tree <strong>of</strong> <strong>human</strong> LINE1elements is called L1Hs (ref. 158); it differs in its 39 untranslatedregion (UTR) by 12 diagnostic substitutions from <strong>the</strong> next oldestsubfamily (L1PA2). Within <strong>the</strong> L1Hs family, <strong>the</strong>re are twosubsets referred to as Ta <strong>and</strong> pre-Ta, de®ned by a diagnostictrinucleotide 193,194 . All active L1 elements are thought to belong to<strong>the</strong>se two subsets, because <strong>the</strong>y account for all 14 known cases <strong>of</strong><strong>human</strong> disease arising from new L1 transposition (with 13 belongingto <strong>the</strong> Ta subset <strong>and</strong> one to <strong>the</strong> pre-Ta subset) 195,196 . Thesesubsets are also <strong>of</strong> great interest for population genetics because atleast 50% are still segregating as polymorphisms in <strong>the</strong> <strong>human</strong>population 194,197 ; <strong>the</strong>y provide powerful markers for tracingpopulation history because <strong>the</strong>y represent unique (non-recurrent<strong>and</strong> non-revertible) genetic events that can be used (along withsimilarly polymorphic Alus) for reconstructing <strong>human</strong> migrations.LINE1 elements that are retrotransposition-competent shouldconsist <strong>of</strong> a full-length sequence <strong>and</strong> should have both ORFs intact.Eleven such elements from <strong>the</strong> Ta subset have been identi®ed,including <strong>the</strong> likely progenitors <strong>of</strong> mutagenic insertions into <strong>the</strong>factor VIII <strong>and</strong> dystrophin genes 198±202 . A cultured cell retrotranspositionassay has revealed that eight <strong>of</strong> <strong>the</strong>se elements remainretrotransposition-competent 200,202,203 .We searched <strong>the</strong> draft <strong>genome</strong> sequence <strong>and</strong> identi®ed 535 LINEsbelonging to <strong>the</strong> Ta subset <strong>and</strong> 415 belonging to <strong>the</strong> pre-Ta subset.These elements provide a large collection <strong>of</strong> tools for probing<strong>human</strong> population history. We also identi®ed those consisting <strong>of</strong>full-length elements with intact ORFs, which are c<strong>and</strong>idate activeLINEs. We found 39 such elements belonging to <strong>the</strong> Ta subset <strong>and</strong>22 belonging to <strong>the</strong> pre-Ta subset; this substantially increases <strong>the</strong>number in <strong>the</strong> ®rst category <strong>and</strong> provides <strong>the</strong> ®rst known examplesin <strong>the</strong> second category. These elements can now be tested forretrotransposition competence in <strong>the</strong> cell culture assay. Preliminary<strong>analysis</strong> resulted in <strong>the</strong> identi®cation <strong>of</strong> two <strong>of</strong> <strong>the</strong>se elements as <strong>the</strong>likely progenitors <strong>of</strong> mutagenic insertions into <strong>the</strong> b-globin <strong>and</strong>RP2 genes (R. Badge <strong>and</strong> J. V. Moran, unpublished data). Similaranalyses should allow <strong>the</strong> identi®cation <strong>of</strong> <strong>the</strong> progenitors <strong>of</strong> most,if not all, o<strong>the</strong>r known mutagenic L1 insertions.L1 elements can carry extra DNA if transcription extends through<strong>the</strong> native transcriptional termination site into ¯anking genomicDNA. This process, termed L1-mediated transduction, provides ameans for <strong>the</strong> mobilization <strong>of</strong> DNA sequences around <strong>the</strong> <strong>genome</strong><strong>and</strong> may be a mechanism for `exon shuf¯ing' 204 . Twenty-one percent <strong>of</strong> <strong>the</strong> 71 full-length L1s analysed contained non-L1-derivedsequences before <strong>the</strong> 39 target-site duplication site, in cases in which<strong>the</strong> site was unambiguously recognizable. The length <strong>of</strong> <strong>the</strong> transducedsequence was 30±970 bp, supporting <strong>the</strong> suggestion that 0.5±1.0% <strong>of</strong> <strong>the</strong> <strong>human</strong> <strong>genome</strong> may have arisen by LINE-basedtransduction <strong>of</strong> 39 ¯anking sequences 205,206 .Our <strong>analysis</strong> also turned up two instances <strong>of</strong> 59 transduction(145 bp <strong>and</strong> 215 bp). Although this possibility had been suggestedon <strong>the</strong> basis <strong>of</strong> cell culture models 195,203 , <strong>the</strong>se are <strong>the</strong> ®rst documentedexamples. Such events may arise from transcription initiatingin a cellular promoter upstream <strong>of</strong> <strong>the</strong> L1 elements. L1transcription is generally con®ned to <strong>the</strong> germline 207,208 , buttranscription from o<strong>the</strong>r promoters could explain a somatic L1retrotransposition event that resulted in colon cancer 206 .Transposons as a creative force. The primary force for <strong>the</strong> origin<strong>and</strong> expansion <strong>of</strong> most transposons has been selection for <strong>the</strong>irability to create progeny, <strong>and</strong> not a selective advantage for <strong>the</strong> host.However, <strong>the</strong>se sel®sh pieces <strong>of</strong> DNA have been responsible forimportant innovations in many <strong>genome</strong>s, for example by contributingregulatory elements <strong>and</strong> even new genes.Twenty <strong>human</strong> genes have been recognized as probably derivedNATURE | VOL 409 | 15 FEBRUARY 2001 | www.nature.com © 2001 Macmillan Magazines Ltd887













