410Luis Franco VeraRev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101en la <strong>que</strong> se proponía “un mo<strong>de</strong>lo plausible” <strong>para</strong>explicar la naturaleza <strong>de</strong> las transiciones alostéricas(13). El mo<strong>de</strong>lo postula <strong>que</strong>, en las proteínasalostéricas, existen dos estados conformacionales, T yR, <strong>que</strong> pue<strong>de</strong>n interconvertirse libremente y <strong>que</strong> presentandistinta afinidad por el ligando, pero no admitela existencia <strong>de</strong> estados intermedios con propieda<strong>de</strong>sestructurales híbridas entre T y R. Los sitios <strong>de</strong> unión<strong>de</strong> ligando a las formas R <strong>son</strong> equivalentes e in<strong>de</strong>pendientesentre sí, y otro tanto ocurre con los sitios <strong>de</strong> lasformas T. Es obvio <strong>que</strong>, si no fuera por la coexistencia<strong>de</strong> T y R, la unión <strong>de</strong> ligando transcurriría sin cooperatividad.Pero cuando se tienen en cuenta los presupuestosanteriores, es evi<strong>de</strong>nte <strong>que</strong>, al añadirligando, éste se unirá preferentemente a las formas <strong>de</strong>afinidad mayor, las formas R. Una simple consi<strong>de</strong>ración<strong>de</strong> las leyes <strong>de</strong>l equilibrio químico indica <strong>que</strong> lapresencia <strong>de</strong> ligando conlleva un <strong>de</strong>splazamiento <strong>de</strong>lequilibrio T-R hacia estas últimas formas, lo <strong>que</strong> dalugar a un incremento global <strong>de</strong> la afinidad. Naturalmente,si se postula <strong>que</strong> los inhibidores alostéricostienen preferencia <strong>para</strong> unirse a las formas T y <strong>que</strong> losactivadores lo hacen predominantemente a las formasR, las mismas consi<strong>de</strong>raciones sobre el equilibrioquímico explican el mecanismo <strong>de</strong> actuación <strong>de</strong> losefectores. Hay <strong>que</strong> señalar, no obstante, <strong>que</strong> estamisma sencillez ha pasado inadvertida a más <strong>de</strong> unautor, <strong>que</strong> se ha limitado a constatar <strong>que</strong> las ecuaciones<strong>de</strong>rivadas <strong>de</strong> ese planteamiento conceptual se ajustancuantitativamente bien al comportamiento experimental<strong>de</strong> la hemoglobina. A título <strong>de</strong> ejemplo, seincluye la ecuación <strong>para</strong> la unión <strong>de</strong>l sustrato:[3]en la <strong>que</strong> es la fracción <strong>de</strong> saturación 8 , L la constantealostérica, es <strong>de</strong>cir la constante <strong>de</strong>l equilibrio T-R en ausencia <strong>de</strong> ligandos, c es la relación entre lasconstantes <strong>de</strong> disociación <strong>de</strong>l sustrato <strong>de</strong> las formas Ty R, y α es el cociente entre la concentración <strong>de</strong> sustratoy su constante <strong>de</strong> disociación <strong>de</strong> las formas R.Poco <strong>de</strong>spués <strong>de</strong> la aparición <strong>de</strong>l artículo <strong>de</strong>Monod, Wyman y Changeux, se publicó otro con elgermen <strong>de</strong> lo <strong>que</strong> sería el segundo gran mo<strong>de</strong>loalostérico, concretamente el <strong>de</strong> Koshland, Némethy yFilmer (14). Para explicar la cooperatividad, Koshlandy sus colaboradores proponían <strong>que</strong> la unión <strong>de</strong> ligandoa una proteína alteraba exclusivamente la conformación<strong>de</strong> la subunidad ocupada. Esto pue<strong>de</strong> afectar ala estabilidad <strong>de</strong> la estructura cuaternaria, si cambia elmodo <strong>de</strong> interacción <strong>de</strong> esa subunidad con las contiguas.Y como la estabilidad pue<strong>de</strong> aumentar o disminuir,pue<strong>de</strong> resultar cooperatividad positiva onegativa. Algo más tar<strong>de</strong>, Koshland extendió elmo<strong>de</strong>lo <strong>para</strong> explicar el alosterismo.Los años siguientes fueron fecundos, ya <strong>que</strong> la publicación<strong>de</strong>l mo<strong>de</strong>lo <strong>de</strong> Koshland inició lo <strong>que</strong>Changeux llamó una “controversia creativa” entre lasdos concepciones <strong>de</strong> cooperatividad y alosterismo. Alser simples y generalizadores, como ha <strong>de</strong> ocurrir contodos los mo<strong>de</strong>los, es ocioso <strong>de</strong>cir <strong>que</strong> prácticamenteninguna proteína alostérica se pue<strong>de</strong> <strong>de</strong>finir al 100%por uno u otro <strong>de</strong> los mo<strong>de</strong>los. Como consecuencia,han surgido mo<strong>de</strong>los alternativos, mo<strong>de</strong>los integradores...,pero en <strong>de</strong>finitiva, cuando han transcurridomás <strong>de</strong> 40 años <strong>de</strong>s<strong>de</strong> la propuesta <strong>de</strong>l alosterismocomo un modo <strong>de</strong> regulación, todos los mo<strong>de</strong>los sereducen <strong>de</strong> una forma u otra a uno u otro <strong>de</strong> los originales.El <strong>de</strong> Monod, Wyman y Changeux tiene elencanto especial <strong>de</strong> las i<strong>de</strong>as geniales, <strong>que</strong> en suaparente sencillez encierran un profundo significado.Por supuesto, es necesario introducir precisionesmatemáticas en las ecuaciones simples <strong>de</strong>l tratamientoinicial, pero el rasgo fundamental <strong>de</strong>l mo<strong>de</strong>lo, es <strong>de</strong>cir,el <strong>que</strong> la transición entre dos estados conformacionalessea concertada, parece resistir los embates <strong>de</strong>l tiempo.Al menos, eso ocurre en el caso <strong>de</strong> la hemoglobina. Enun reciente estudio exhaustivo, se han analizado datoscinéticos muy precisos sobre la disociación <strong>de</strong> complejoshemoglobina-monóxido <strong>de</strong> carbono <strong>para</strong> concluir<strong>que</strong> el mo<strong>de</strong>lo <strong>de</strong> dos estados sirve <strong>para</strong> explicarsatisfactoriamente la cooperatividad en la unión <strong>de</strong> ligandosa la hemoglobina (15). Es necesario, eso sí, unsistema <strong>de</strong> 85 ecuaciones diferenciales acopladas <strong>para</strong><strong>de</strong>scribir a<strong>de</strong>cuadamente los resultados <strong>de</strong>s<strong>de</strong> un punto<strong>de</strong> vista cuantitativo, pero el rasgo fundamental <strong>de</strong>lmo<strong>de</strong>lo ha persistido.8 La fracción <strong>de</strong> saturación es la fracción molar <strong>de</strong> sitios ocupados por ligando.
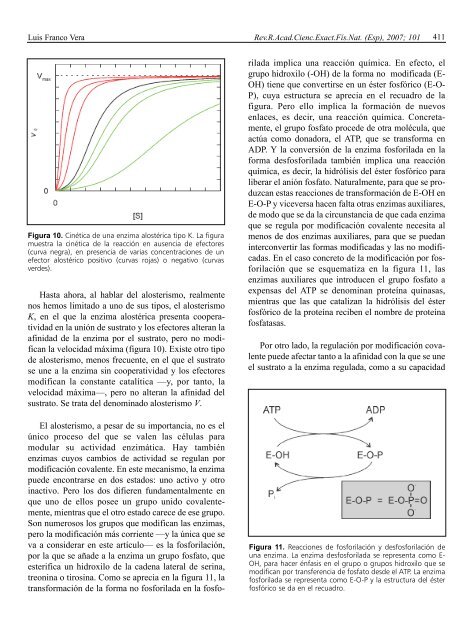
Luis Franco Vera Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp), 2007; 101 411Figura 10. Cinética <strong>de</strong> una enzima alostérica tipo K. La figuramuestra la cinética <strong>de</strong> la reacción en ausencia <strong>de</strong> efectores(curva negra), en presencia <strong>de</strong> varias concentraciones <strong>de</strong> unefector alostérico positivo (curvas rojas) o negativo (curvasver<strong>de</strong>s).Hasta ahora, al hablar <strong>de</strong>l alosterismo, realmentenos hemos limitado a uno <strong>de</strong> sus tipos, el alosterismoK, en el <strong>que</strong> la enzima alostérica presenta cooperativida<strong>de</strong>n la unión <strong>de</strong> sustrato y los efectores alteran laafinidad <strong>de</strong> la enzima por el sustrato, pero no modificanla velocidad máxima (figura 10). Existe otro tipo<strong>de</strong> alosterismo, menos frecuente, en el <strong>que</strong> el sustratose une a la enzima sin cooperatividad y los efectoresmodifican la constante catalítica —y, por tanto, lavelocidad máxima—, pero no alteran la afinidad <strong>de</strong>lsustrato. Se trata <strong>de</strong>l <strong>de</strong>nominado alosterismo V.El alosterismo, a pesar <strong>de</strong> su importancia, no es elúnico proceso <strong>de</strong>l <strong>que</strong> se valen las células <strong>para</strong>modular su actividad enzimática. Hay también<strong>enzimas</strong> cuyos cambios <strong>de</strong> actividad se regulan pormodificación covalente. En este mecanismo, la enzimapue<strong>de</strong> encontrarse en dos estados: uno activo y otroinactivo. Pero los dos difieren fundamentalmente en<strong>que</strong> uno <strong>de</strong> ellos posee un grupo unido covalentemente,mientras <strong>que</strong> el otro estado carece <strong>de</strong> ese grupo.Son numerosos los grupos <strong>que</strong> modifican las <strong>enzimas</strong>,pero la modificación más corriente —y la única <strong>que</strong> seva a consi<strong>de</strong>rar en este artículo— es la fosforilación,por la <strong>que</strong> se aña<strong>de</strong> a la enzima un grupo fosfato, <strong>que</strong>esterifica un hidroxilo <strong>de</strong> la ca<strong>de</strong>na lateral <strong>de</strong> serina,treonina o tirosina. Como se aprecia en la figura 11, latransformación <strong>de</strong> la forma no fosforilada en la fosforiladaimplica una reacción química. En efecto, elgrupo hidroxilo (-OH) <strong>de</strong> la forma no modificada (E-OH) tiene <strong>que</strong> convertirse en un éster fosfórico (E-O-P), cuya estructura se aprecia en el recuadro <strong>de</strong> lafigura. Pero ello implica la formación <strong>de</strong> nuevosenlaces, es <strong>de</strong>cir, una reacción química. Concretamente,el grupo fosfato proce<strong>de</strong> <strong>de</strong> otra molécula, <strong>que</strong>actúa como donadora, el ATP, <strong>que</strong> se transforma enADP. Y la conversión <strong>de</strong> la enzima fosforilada en laforma <strong>de</strong>sfosforilada también implica una reacciónquímica, es <strong>de</strong>cir, la hidrólisis <strong>de</strong>l éster fosfórico <strong>para</strong>liberar el anión fosfato. Naturalmente, <strong>para</strong> <strong>que</strong> se produzcanestas reacciones <strong>de</strong> transformación <strong>de</strong> E-OH enE-O-P y viceversa hacen falta otras <strong>enzimas</strong> auxiliares,<strong>de</strong> modo <strong>que</strong> se da la circunstancia <strong>de</strong> <strong>que</strong> cada enzima<strong>que</strong> se regula por modificación covalente necesita almenos <strong>de</strong> dos <strong>enzimas</strong> auxiliares, <strong>para</strong> <strong>que</strong> se puedaninterconvertir las formas modificadas y las no modificadas.En el caso concreto <strong>de</strong> la modificación por fosforilación<strong>que</strong> se es<strong>que</strong>matiza en la figura 11, las<strong>enzimas</strong> auxiliares <strong>que</strong> introducen el grupo fosfato aexpensas <strong>de</strong>l ATP se <strong>de</strong>nominan proteína quinasas,mientras <strong>que</strong> las <strong>que</strong> catalizan la hidrólisis <strong>de</strong>l ésterfosfórico <strong>de</strong> la proteína reciben el nombre <strong>de</strong> proteínafosfatasas.Por otro lado, la regulación por modificación covalentepue<strong>de</strong> afectar tanto a la afinidad con la <strong>que</strong> se uneel sustrato a la enzima regulada, como a su capacidadFigura 11. Reacciones <strong>de</strong> fosforilación y <strong>de</strong>sfosforilación <strong>de</strong>una enzima. La enzima <strong>de</strong>sfosforilada se representa como E-OH, <strong>para</strong> hacer énfasis en el grupo o grupos hidroxilo <strong>que</strong> semodifican por transferencia <strong>de</strong> fosfato <strong>de</strong>s<strong>de</strong> el ATP. La enzimafosforilada se representa como E-O-P y la estructura <strong>de</strong>l ésterfosfórico se da en el recuadro.